The 2nd Big Data Forum for Life and Health Sciences
The 2nd Big Data Forum for Life and Health Sciences (October 10-13, 2017)
Biological research has entered the era of big data, including a wide variety of omics data and covering a broad range of health data. Such big data is generated at ever-growing rates and distributed throughout the world with heterogeneous standards and diverse limited access capabilities. However, the promise to translate these big data into big knowledge can be realized only if they are publicly shared. Thus, providing open access to omics & health big data is essential for expedited translation of big data into big knowledge and is becoming increasingly vital in advancing scientific research and promoting human healthcare and precise medical treatment.
Share Big Data, Make Big Discovery
It is our great pleasure to announce that the 2017 Big Data Forum for Life and Health Sciences will be held in October 10-13, 2017. A few renowned biomedical data scientists have agreed to give speeches. Likely, you are also cordially invited to share your work and participate in this excited event.
Looking forward to seeing you in Beijing, China! We will be working hard to ensure your stay not only a fruitful one, but also an enjoyable one!
会议通知下载
{kind=link}

Invited Speakers

Amir Ali Abbasi
Professor
National Center for Bioinformatics
Quaid-i-Azam University
Pakistan

Shahid Mahmood Baig
Professor of Human Molecular Genetics
Head of Health Biotechnology Division
National Institute for Biotechnology and Genetic Engineering
Pakistan

Yiming Bao
Professor
Director of BIG Data Center
Beijing Institute of Genomics (BIG), CAS

Alex Bateman
Professor
Head of Protein Sequence Resources
EMBL-EBI
United Kingdom

Ling-Ling Chen
Professor
College of Informatics
Huazhong Agricultural University
China

Runsheng Chen
Professor, Member of the CAS
Principal Investigator
Institute of Biophysics, CAS

Mikhail S. Gelfand
Professor
Vice-Director for ITTP Science
Head of IITP Research and Training Center of Bioinformatics
Russia Academy of Sciences

Songnian Hu
Professor
Director of Key Laboratory of Genome Sciences & Information
Beijing Institute of Genomics, CAS

Dong Li
Professor
Laboratory of Biological Networks
National Center for Protein Sciences · Beijing
China

Jiao Li
Professor
Institute of Medical Information and Library
Chinese Academy of Medical Sciences

Lei Li
Professor
Academy of Mathematics and Systems Science
CAS

Jiang Liu
Professor
Associate Director of Key Laboratory of Genome Sciences & Information
Beijing Institute of Genomics, CAS

Lei Liu
Professor
Institute of Biomedical Sciences
Fudan University
China

Weizhong Li
Professor
Zhongshan School of Medicine
Sun Yat-Sen University
China

Yasukazu Nakamura
Professor, Head of Database Division
DDBJ Center, National Institute of Genetics
Japan

Etienne PAUX
Head of Database Division
National Institute of Agricultural Research
France

Bairong Shen
Professor
Director of Center for Systems Biology
Soochow University
China

Chris Sander
Professor of Cell Biology at Harvard Medical School
Director of the cBio Center at the Dana-Farber Cancer Institute
Associate Member of the Broad Institute
United States

Dr. William Summerskill
Senior Executive Editor
The Lancet

Vorasuk Shotelersuk
Professor, Associate Dean for Research Affairs
Director of Excellence Center for Medical Genetics
Faculty of Medicine, Chulalongkorn University
Thailand

Qianfei Wang
Professor
Associate Director of Key Laboratory of Genomic and Precision Medicine
Beijing Institute of Genomics, CAS

Xiujie Wang
Professor
Director of Center for Molecular Systems Biology
Institute of Genetics and Developmental Biology, CAS

Shuhua Xu
Professor
Human Population Genetics
CAS-MPG Partner Institute for Computational Biology (PICB)

Yu Xue
Professor
College of Life Science and Technology
Huazhong University of Science and Technology
China

Fangqing Zhao
Professor
Beijing Institutes of Life Science
CAS

Wenming Zhao
Associate Director of BIG data center
Beijing Institute of Genomics (BIG), CAS

Yongzhen Zhang
Professor
National institute for communicable disease control and prevention
Chinese center for disease control and prevention
China
Agenda
| October 10: Pick-up & Registration | |
|---|---|
| October 11: Talks | |
| 08:50 - 10:00 | Session 1, chaired by Zhang Zhang, Beijing Institute of Genomics (BIG), CAS |
| 08:50 - 09:00 | Welcome and Opening Remarks Dr. De-Xing Zhang, Professor Deputy Director of Beijing Institute of Genomics (BIG), CAS |
| 09:00 - 09:30 |
BIG Data Center: Current Status and Future Directions Yiming Bao, Beijing Institute of Genomics (BIG), CAS |
| 09:30 - 10:00 |
High incidence of inherited diseases in inbred Pakistani population: Use of genomic and
bioinformatic tools for molecular characterization [Abstract]
Shahid Mahmood Baig, National Institute for Biotechnology and Genetic Engineering, Pakistan |
| Pakistan has the highest incidence of genetic diseases due to strong local tradition of consanguineous marriages and large family size in this country. β-thalassemia is the most common single gene genetic disorder in this country with>6% allele frequency. Similarly, the incidence of inherited hearing impairment, primary microcephaly (MCPH) and other disorders is higher than other populations of the world. Identification of the genes underlying these disorders in the inbred Pakistani population is carried out using modern genomic techniques at Human Molecular Genetics Laboratory (HMGL), National Institute for Biotechnology and Genetic Engineering (NIBGE) to establish genetic counseling, carrier screening, prenatal diagnosis (PND) for prevention, improved treatment and personalized medicine of inherited diseases. In the framework of this program, we analyzed more than 1600 β-thalassemia families through cascade testing and offered first trimester PND through chorionic villus sampling (CVS) to carrier couples. In the last 8 years >1100 retrospective PND of β-thalassemia have been performed. A family with glioblastoma (brain tumor) was also characterized at molecular level and DNA based first trimester PND diagnoses were provided in three at risk pregnancies. In addition, more than 2200 large consanguineous families with various disease phenotypes [e.g. Primary microcephaly (MCPH), Neurodevelopmental disorders, hearing impairment, eye disorders, Ectodermal dysplasias, Intellectual disability, peripheral neuropathies, skeletal and limb deformities etc.) having at least two affected individuals were identified through field sampling trips in diverse regions of Pakistan and analyzed for identification of disease genes. In case of MCPH, we have reported CEP135, CDK6 , and PLK4 as the disease causing genes of MCPH. We recently identified homozygous mutations in KIF14 (NM_014875.2;c.263T>A;pLeu88*, c.2480_2482delTTG;p.Val827del and c.4071G>A;p.Gln1357=) as the likely cause in three MCPH families. Further, in a patient presenting with a severe form of primary microcephaly and short stature, we identified compound heterozygous missense mutations in KIF14(NM_014875.2;c.2545C>G;p.His849Asp and c.3662G>T;p.Gly1221Val). Three of the five identified mutations impaired splicing and two resulted in a truncated protein. Intriguingly, Kif14 knockout mice also showed primary microcephaly. Human kinesin-like protein KIF14, a microtubule motor protein, localizes at the midbody to finalize cytokinesis by interacting with CRIK. We found impaired localization of both KIF14 and CRIK at the midbody in patient-derived fibroblasts. Further, we observed a large number of binucleated and apoptotic cells — signs of failed cytokinesis that we also observed in experimentally KIF14-depleted cells. Our data corroborate the role of an impaired cytokinesis for the etiology of primary and syndromic microcephaly as has been proposed by recent findings on CIT mutations. A number of novel disease loci, genes and mutations have been identified by HMGL for various monogenic disorders in collaboration with various research groups in Europe, US and China. The gene hunting for several genetic diseases is in progress under the framework of international collaborative projects and we are optimistic to characterize the molecular basis of several diseases and handling of huge genomic data generated for providing the important insights for developing efficient disease prevention and therapeutic approaches. | |
| 10:00 - 10:20 | Tea & Coffee Break |
| 10:20 - 12:00 | Session 2, chaired by Yiming Bao, Beijing Institute of Genomics (BIG), CAS |
| 10:20 - 10:50 |
Healthcare and Biomedical Big Data Management
[Abstract]
Bairong Shen, Soochow University |
| With the advances of big data technologies and precision medicine, healthcare is becoming the next frontier both for scientific research and economic development. I will review the big healthcare data management such as, collection and analysis; standardization and ontology, personal data privacy and systems level modeling for the understanding and intervention of health. I will emphasize the development of physiological informatics which will be the focus of health management research. The physiological data from wearable sensors and intelligent mobile phone is rapidly accumulated, which provides us the opportunity to collect the dynamic and personalized data and integrated them into genotype information to fully understand the complex health and disease evolution. This can be applied to the early prediction and prevention of disease and promote the paradigm shifting from treatment of disease to healthcare. | |
| 10:50 - 11:20 |
Mining free texts towards biomedical knowledge discovery: from literature to EHR
[Abstract]
Jiao Li, Institute of Medical Information and Library, Chinese Academy of Medical Sciences |
| Biomedical scientific studies were reported and communicated via scientific literature. Meanwhile, clinical observations and therapeutic procedures were reported in electronic health records (EHR). Both literature and EHR consist of meaningful and unstructured free texts, thus, text mining becomes increasingly important in biomedical research by extracting key information from free texts and converting it into structured knowledge. In this talk, I will introduce our text mining efforts applied in computational drug repositioning, precision medicine ontology construction, clinical pharmacogenomics information identification as well as Chinese symptom identification and standardization. | |
| 11:20 - 12:00 |
The quality continuum: from protocol to publication [Abstract] William Summerskill, The Lancet |
| Publishing top quality research is not the result of special tricks in writing a paper. High quality publication begins with asking the right question, in the right way, at the right time. Then following the right methods to ensure that the data will be publishable and able to inform care. This presentation will show ways to strengthen clinical research and improve the likelihood of publishing in your preferred journal. | |
| 12:00 - 13:30 | Lunch |
| 13:30 - 15:00 | Session 3, chaired by Lei Liu, Institute of Biomedical Sciences, Fudan University |
| 13:30 - 14:00 |
Clinical NGS for Rare and Undiagnosed Diseases
[Abstract]
Vorasuk Shotelersuk, Faculty of Medicine, Chulalongkorn University, Thailand |
|
Rare diseases, each defined by its prevalence of fewer than 1 in 2,000 people, collectively accounts
for as much as 8% of the population (EURORDIS, 2005). At least 7,000 distinct rare diseases exist,
about 80% have a genetic component, and only around 400 rare diseases currently have therapies.
Patients with rare diseases typically endure around 7 years of investigations before their diagnosis
is confirmed. Molecular techniques have rapidly advanced in recent years. They have played an important role not only in researches, but also medical practice. I will present our experience in using NGS to revolutionize a molecular diagnostic method. Etiologic mutations of rare genetic disorders including those with many possible causative genes such as skeletal dysplasia, congenital heart diseases, intellectual disabilities and epileptic encephalopathy can be efficiently identified by whole exome sequencing (WES) or whole genome sequencing (WGS). To date, we has completed over 1000 WES in over 50 different diseases. This has provided a tremendous diagnostic rate of 40% leading to the discovery of 4 new disease genes (Nat Commun:2016;6;7:11920). With definite diagnoses and classification of diseases by its mutated genes, a more specific management guideline for that particular patient and a more precise genetic counselling can be given. This also leads to new and hopeful preventive and treatment strategies. |
|
| 14:00 - 14:30 |
Computational analysis of PTMs in autophagy
[Abstract]
Yu Xue, Huazhong University of Science and Technology |
| Autophagy is a highly conserved process for degrading cytoplasmic contents, determines cell survival or death, and regulates the cellular homeostasis. Besides core ATG proteins, numerous regulators together with various post-translational modifications (PTMs) are also involved in autophagy. Recent studies demonstrated that the dysregulation of macroautophagy/autophagy is involved in human diseases such as cancers and neurodegenerative disorders. Thus, autophagy has become a promising therapeutic target for biomedical design. Here, we developed a database of The Autophagy, Necrosis, ApopTosis OrchestratorS (THANATOS, http://thanatos.biocuckoo.org), containing 191,543 proteins potentially associated with autophagy and cell death pathways in 164 eukaryotes. We performed an evolutionary analysis of core ATG genes, and observed that ATGs required for the autophagosome formation are highly conserved across eukaryotes. Further analyses revealed that known cancer genes and drug targets were over-represented in human autophagy proteins, which were significantly associated in a number of signaling pathways and human diseases. By re-constructing a human kinase-substrate phosphorylation network for core ATG proteins, our results confirmed that phosphorylation play a critical role in regulating autophagy. Using this data resource, we performed a quantitative phosphoproteomic profiling to delineate the phosphorylation signalling networks regulated by 2 natural neuroprotective autophagy enhancers, corynoxine (Cory) and corynoxine B (Cory B). We developed a novel algorithm of in silico Kinome Activity Profiling (iKAP) to predict that Cory or Cory B potentially regulates different kinases. We discovered 2 kinases, MAP2K2/MEK2 (mitogen-activated protein kinase kinase 2) and PLK1 (polo-like kinase 1), to be potentially upregulated by Cory, whereas the siRNA-mediated knockdown of Map2k2 and Plk1 significantly inhibited Cory-induced autophagy. Furthermore, Cory promoted the clearance of Alzheimer disease-associated APP (amyloid beta [A4] precursor protein) and Parkinson disease-associated synuclein alpha (SNCA/α-synuclein) by enhancing autophagy, and these effects were dramatically diminished by the inhibition of the kinase activities of MAP2K2 and PLK1. Taken together, our study not only provided bioinformatics resources and approaches for analyzing PTMs in autophagy, but also identified the important role of MAP2K2 and PLK1 in neuronal autophagy. | |
| 14:30 - 15:00 |
Diversified RNA-level regulation in controlling the pluripotency of
embryonic stem cells
[Abstract] Xiujie Wang, Institute of Genetics and Developmental Biology, CAS |
| The pluripotency of embryonic stem cells (ESCs) is regulated by multiple layers of factors. At the RNA level, we have identified a cluster of miRNAs whose expression abundance is positively correlated with the pluripotency level of ESCs, and confirmed that one of the functions of these miRNAs is to target the key component of the PRC2 complex therefore to regulate H3K27me3 modification. We have proven that ESC-specific transcription factors are capable to produce ESC-specific transcripts with alternative transcription start sites from ubiquitously expressed genes, thus confer ubiquitously expressed genes novel functions to involve in the maintenance of ESC pluripotency. In addition, the m6A modification on mRNAs is also required for reprogramming of MEFs to induced pluripotency stem cells (iPSCs). | |
| 15:00 - 15:30 | Tea & Coffee Break |
| 15:30 - 17:00 | Session 4, chaired by Yu Xue, Huazhong University of Science and Technology |
| 15:30 - 16:00 |
DDBJ and the INSDC
[Abstract] Yasukazu Nakamura, DDBJ Center, National Institute of Genetics, Japan |
| Since 1987, the DNA Data Bank of Japan (DDBJ) at the National Institute for Genetics in Japan has worked as a partner of the International Nucleotide Sequence Database Collaboration (INSDC). The INSDC has been committed to capturing, preserving and providing access to comprehensive public domain nucleotide sequence and associated metadata which enables discovery in biomedicine, biodiversity and biological sciences. We have worked collaboratively to enable access to nucleotide sequence data in standardized formats for the worldwide scientific community. In this talk, I will present DDBJ’s activities and the principles of the INSDC collaboration, and summarize the trends of the nucleotide archival contents. | |
| 16:00 - 16:30 |
Building Knowledgebase for Precision Medicine [Abstract]
Lei Liu, Institute of Biomedical Sciences, Fudan University |
| With the development of science and technology has entered intelligent era, Biomedicine will become more intelligent and precise. The trend of precision medicine study is the integration of various new technologies including big data technology, omics technology, bioinformatics, and so on. The research in life sciences has gradually turned into the mode of data-driven discovery. The key step in the process is the understanding and annotating the biomedical big data. We believe that the integrated analysis of clinical and bioinformatics data can make breakthrough in precision medicine. Precision medicine will revolutionize the medical practice, allow doctors make personalized and precise diagnosis, treatment and prevention plan based on patients' personal genomic data, life style and other specific factors. | |
| 16:30 - 17:00 |
Reprogramming of epigenetic landscape during embryo development Jiang Liu, Beijing Institute of Genomics (BIG), CAS |
| 18:00 - 20:00 | Welcome Dinner |
| October 12: Talks | |
| 9:00 - 10:10 | Session 5, chaired by Wenming Zhao, Beijing Institute of Genomics (BIG), CAS |
| 09:00 - 09:40 |
UniProt: A universal hub of protein knowledge [Abstract]
Alex Bateman, EMBL-EBI, United Kingdom |
| In this presentation I will discuss the UniProt KnowledgeBase which has been providing data for protein sequence and function for over 30 years. I will discuss how we are dealing with the deluge of sequence data to organise it in ways to make our users lives easier and more scalable. I will show with examples how our expert curators gather knowledge from the literature, evaluate it and add value through structuring it with a variety of ontologies. Does expert curation scale with the vast biomedical literature? I will try to address this important point. Firstly through recent experiments to estimate the fraction of the literature we curate and secondly through automated systems to annotate proteins that have never been experimentally characterised. Finally I will discuss how UniProt helps to connect and integrate the worldwide community of biological resources and their data. | |
| 09:40 - 10:10 |
Rice Information GataWay (RIGW) and CRISPR-P genome editing tool in plants [Abstract] Lingling Chen, Huazhong Agricultural University |
|
We established a comprehensive bioinformatics platform RIGW to provide GBrowse-based view of ZS97
and MH63 genomic and other omics data. RIGW offered homologs among indica and japonica rice, and
other plant species. We also provided user-friendly web interfaces to show the predicted PPIs in
rice, the metabolic pathways in ZS97/MH63, CRISPR-Cas single guide RNA design tool and GO enrichment
in RIGW. All the genomic sequences and annotation could be freely accessed, and useful links to
other public databases were offered. RIGW is freely available at http://rice.hzau.edu.cn/. CRISPR-P provides web services for computer-aided design of sgRNA with minimal off-target potentials, which supports sgRNA design for most of the sequenced plant genomes. CRISPR-P supports to design guide sequences for various CRISPR-Cas systems including Cpf1 and various Cas9 endonucleases. A comprehensive analysis of the guide sequence is provided, including GC content, restriction endonuclease site, microhomology sequence flanking the targeting site (microhomology score), and the secondary structure of sgRNA. Identification of sgRNA from custom sequences is also provided. CRISPR-P is freely available at http://cbi.hzau.edu.cn or http://cbi.hzau.edu.cn/CRISPR2/. |
|
| 10:10 - 10:30 | Tea & Coffee Break |
| 10:10 - 12:00 | Session 6, chaired by Lingling Chen, Huazhong Agricultural University |
| 10:30 - 11:00 |
A Dual Eigen-Analysis of Mouse Multi-Tissues’ Expression Profiles Unveils New Perspectives
into Type 2 Diabetes Lei Li, Academy of Mathematics and Systems Science, CAS |
| 11:00 - 11:30 |
Three-dimensional structure and functional states of chromatin
[Abstract]
Mikhail S. Gelfand, IITP Research and Training Center of Bioinformatics, Russia Academy of Sciences, Russia |
| Recent advances in experimental methods create an opportunity to integrate data about contacts between DNA regions, their epigenetic state, and gene expression. At the small scale, chromatin forms relatively tight globules, so called Topologically Associating Domains. In Drosophila, housekeeping and highly transcribed genes are enriched in inter-TAD regions, whereas tissue-specific and silent genes tend to be localized in TADs. Inter-TAD regions are enriched in active histone marks, whereas TADs are mainly formed by repressed chromatin. Moreover, differences in TAD structure between cell lines are accompanied by changes in expression of genes situated in such TADs. On the other hand, unlike mammals, TAD boundaries in Drosophila are not highly enriched in binding sites for insulator proteins such as CTCF. Similar trends are also observed in unicellular eukaryote Dictyostelium discoideum. | |
| 11:30 - 12:00 |
Can Genomics Guide Cancer Treatment? A Tale of Aggressive NK-cell Leukemia
Qianfei Wang, Beijing Institute of Genomics (BIG), CAS |
| 12:00 - 13:30 | Lunch |
| 13:30 - 15:10 | Session 7, chaired by Yiming Bao, Beijing Institute of Genomics (BIG), CAS |
| 13:30 - 14:10 |
Genomics, Big Data and Precision Medicine Runsheng Chen, Institute of Biophysics, CAS |
| 14:10 - 14:40 |
A bioinformatics platform system for omics data analysis at Sun Yat-sen University
[Abstract]
Weizhong Li, Sun Yat-Sen University |
| We establish a bioinformatics platform system (http://lilab.sysu.edu.cn/Tools/) for omics data analysis at Sun Yat-sen University, which currently offers applications of sequence similarity search (SSS), genomic data workflows (GDW), deep learning analysis (DLA), multiple sequence alignment (MSA) and protein functional analysis (PFA). Biological sequence data including UniProt, ENA/GenBank and 1000Genomes are searchable through our PSISearch2, BLAST and FASTA applications; gene variations and protein binding sites can be functionally analysed by the deep learning applications DeepSea and DeepBind; Clustal Omega and InterProScan5 are integrated for multiple sequence alignment and protein functional analysis. Ongoing implementations include latest genomic analysis workflows for whole exome sequencing, whole genome sequencing and RNA-seq data, and the ultrafast TB-scale genomic data search tools - SBTblast & ReverseSearch. Analysis jobs can be run through webform interfaces and web service APIs. Example web service client programs in common computing languages such as Java, Perl & Python are provided for user to run high-throughput analyses systematically and consume our computing resource remotely. To facilitate software and data sharing, the platform system can be packed by Docker container and migrated to other high-performance computing clusters. | |
| 14:40 - 15:10 |
Understanding Human Genetic History and Local Adaptation with Big Genomic Data [Abstract]
Shuhua Xu, CAS-MPG Partner Institute for Computational Biology (PICB) |
| Recent advances in genotyping and sequencing technologies have facilitated genome-wide investigation of human genetic variations and provided new insights into population history and genotype-phenotype relationships. Here I will present some of our recent progresses in studying human genetic history and local adaptation with large-scale population genomic data. | |
| 15:10 - 15:30 | Tea & Coffee Break |
| 15:30 - 17:00 | Session 8, chaired by Shuhua Xu, CAS-MPG Partner Institute for Computational Biology (PICB) |
| 15:30 - 16:00 |
High diversity of viruses in nature - new challenges in the prevention and control of
infectious diseases Yongzhen Zhang, Chinese center for disease control and prevention |
| 16:00 - 16:30 |
Computational methods in exploring circular noncoding RNAs Fangqing Zhao, Beijing Institutes of Life Science, CAS |
| 16:30 - 17:00 |
GSA: Genome Sequence Archive Wenming Zhao, Beijing Institute of Genomics (BIG), CAS |
| October 13: Talks | |
| 09:00 - 10:10 | Session 9, chaired by Jingchu Luo, Peking University |
| 09:00 - 09:40 |
Solving Hard Life Science Problems:
1. Prediction of 3D Structures of Proteins, RNAs, and Molecular Complexes 2. Design of Cancer Combination Therapy [Abstract] Chris Sander, Dana-Farber Cancer Institute, Harvard University, United States |
| Part 1: Collaborative efforts combining computational biology, structural biology and statistical physics expertise provide a solution to the computational protein folding problem. Genomic sequences contain rich evolutionary information about functional constraints on macromolecules such as proteins. This information can be efficiently mined to detect evolutionary couplings between residues in proteins and address the longstanding challenge to compute protein and RNA three dimensional structures from sequences alone. Substantial progress on the evolutionary couplings approach, since the initial attempts in 1994, has become possible because of the explosive growth in available sequences and the application of global statistical methods, such as maximum entropy distillation of correlated mutation patterns. In addition to proteins and RNA 3D structure, this powerful analysis of covariation helps identify functional residues involved in ligand binding, complex formation and conformational changes. We expect computation of evolutionary covariation patterns to help elucidate the full spectrum of protein and RNA structures, their functional interactions and evolutionary dynamics. Collaboration between the Sander and Marks (Harvard Medical School) groups, as well as initially with Martin Weigt, Andrea Pagnani and Riccardo Zecchina at Politecnico di Torino. Use the http://evfold.org server to compute EVcouplings and to predict 3D structure for large sequence families. Ref: Protein 3D Structure from high-throughput sequencing http://bit.ly/tob48p . Ref: 3D RNA and Functional Interactions from Evolutionary Couplings http://bit.ly/3D_RNA . Part 2: Cells and organisms have evolved as robust to external perturbations and adaptable to changing conditions. This capacity poses severe problems for cancer patients. Some targeted anti-cancer drugs work remarkably well, yet resistance is almost certain to emerge. Three particular scientific challenges arise: (1) discover the escape routes in response to drugs and how to block the exits by combinatorial intervention; (2) in The Cancer Genome Atlas empirically describe the landscape of oncogenic alterations for improved therapeutic navigation and (3) use experimental perturbation biology (systematic perturbation coupled with rich observation of response, such as changes in protein levels and protein modifications) to derive executable network models for cancer cells that guide the development of combination therapy. Work done in collaboration with Anil Korkut, Evan Molinelli, Martin Miller, Wei Qing Wang, Xiaohong Jing, Alex Root, Deb Bemis, David Solit, Christine Pratilas, Emek Demir, Arman Aksoy, Onur Sumer, Özgün Babur, Andrea Pagnani, Martin Weigt, Riccardo Zecchina, Giovanni Ciriello, Nikolaus Schultz, Sven Nelander, Debora Marks et al. Ref: Perturbation biology nominates upstream– downstream drug combinations in RAF inhibitor resistant melanoma cells http://bit.ly/2cB4jNv. | |
| 09:40 - 10:10 |
Improving metazoans non-coding genome annotation in BIG DATA context
[Abstract]
Amir Ali Abbasi, Quaid-i-Azam University, Pakistan |
| The completion of the human genome sequence was a major landmark event in the history of modern biology. The next challenge was to decode and characterize the entire repertoire of functional elements relevant to human biology and disease. Early genome annotations attempts quickly revealed that ~98.5% of our DNA do not code for proteins. Now the question was whether the remaining large fraction of noncoding human genome is useless junk or it encodes (at least subset) for unknown functions? Comparative developmental and genetic studies between various animal lineages have implicated noncoding metazoans DNA in macro-and microevolutionary processes. More recently, genome-wide association studies have indicated that a large majority of trait/disease-associated genomic segments lie in noncoding portion of our genome. These findings create a great deal of interest to identify and characterize all noncoding functional elements in the human genome. This talk would highlight, how the DNA sequencing of hundreds of animal genomes and cheaper in vitro assays has allowed the functional interrogation of noncoding human genome, based on sequence conservation or irrespective of sequence conservation. Furthermore, this talk would highlight the relative interpretation, merits and caveats of the various high-throughput approaches currently in use to decode the noncoding functional segments in the human genome. | |
| 10:10 - 10:30 | Tea & Coffee Break |
| 10:30 - 12:00 | Session 10, chaired by Jingfa Xiao, Beijing Institute of Genomics (BIG), CAS |
| 10:30 - 11:00 |
Knowledge mining from the large scale protein-protein interaction networks
[Abstract]
Dong Li, National Center for Protein Sciences · Beijing |
|
Throughout the history of natural science, it is definite that our knowledge and disciplines come
from accumulated discoveries, which are triggered by the unprecedented scale and speed of big data
and achieved by efficient mathematical strategies, taking the discovery of Kepler’s laws as an
example. In my laboratories, simple machine learning strategies, such as Naïve Bayesian network, have been used to find the instinct features of proteome organization, especially the protein interactions. By using naïve Bayesian network, reliability was assigned to the human protein-protein interactions identified by high throughput experiments by combining multiple heterogeneous biological evidences. Then domain enrichment ratio was introduced to measure the direction between interacting proteins, resulting in an integrated human directional protein interaction network. Next, logistic regression was taken to integrate six representative features, to develop a proteome-wide prediction model of self-interacting proteins. Recently, we developed a naïve Bayesian classifier to combine multiple heterogeneous biological evidences to investigate the human E3-substrate interactions which determine the high specificity of ubiquitination. UbiBrowser is now provided as an integrated bioinformatics platform to predict and present the proteome-wide human E3-substrate interaction network. It is believed that the era of big data will bring in new insights in life sciences and present new opportunities in research. Artificial intelligence strategy will play dominant roles in the coming knowledge discovery. My colleagues are now engaged to develop an automatic knowledge discovery highway(ProDiGy), integrating the OMICS datasets and literature information into a biomedical knowledge graph (a heterogamous information network) followed by the feature extraction and deep learning for grand knowledge. |
|
| 11:00 - 11:30 |
The big wheat genome in the big data era [Abstract]
Etienne Paux, National Institute of Agricultural Research, France |
| Wheat is one of the most important crops worldwide, providing food for ~30% of the world population. While significant advances have been made since the Green Revolution, today’s agriculture is facing unprecedented challenges: to keep pace with the growing human demand in an environmentally and socially sustainable manner and in a context of climate change. It has long been argued that a better knowledge of the wheat genome should help to face this challenge. However, because of its size, allohexaploid nature, and high repeat content, the wheat genome has long been perceived as too complex for molecular studies. The advent of high-throughput genotyping and sequencing technologies have opened new perspectives and led to a paradigm change. As a result, the International Wheat Genome Sequencing Consortium (IWGSC) has recently achieved a high-quality reference sequence of the wheat genome that has been analyzed in details through the integration of several genomics data. These analyses revealed the wheat genome peculiarities, such as an uneven distribution of recombination events, a strong structural and functional partitioning, a higher proportion of non-syntenic and duplicated genes compared to other crops, a correlation between gene and genome structure and expression as well as the existence of chromosomal domains enriched in co-expressed genes. These characteristics are the result of wheat evolution. However, compared to wild species, wheat has a complex history of evolution that also includes domestication and breeding. Results on the wheat genome structure, function and evolution will be presented, together with examples illustrating the intricate links between the wheat genome organization and wheat breeding, and showing how one can impact the other. | |
| 11:30 - 12:00 |
New sequencing technology and assembly tools for genome of plant species
[Abstract]
Songnian Hu, Beijing Institute of Genomics,CAS |
|
With the advance of NGS technology, more and more complex genome were decoded in last decade. Plant
genomes are often with large size, high heterozygosity, or with recent whole genome duplication
events/allotetraploid events, due to the large gene family, recent tandem gene duplication and
cluster located repeats. Compared to other species genomes, the assemble quality of plant genome was
far below average, and made the protein prediction, genome companion and selective sweep detection
more difficult and inaccuracy.
The emergence of third generation sequencing platform and other chromosome level detection methods in recent years offered a good solution for the plant genome assembly. Now, the route solution for de novo plant genome sequencing project consists of those steps: NGS assembly/PacBio assembly, scaffolding by HiC-seq and/or 10X genomic,improvement by BioNano/optical map, and anchoring the superscaffolds to chromosome by high density genetic map. Practically, NGS assembly for genome with high heterozygosity, PacBio assembly for high complexity genome with high heterozygosity or large genome size could be heavily affected by software and parameter selection. Careful software selection and comparisons should be carried out for sequencing based assembly under the guidance of BioNano or genetic map. |
|
| 12:00 - 12:10 | Closing Remarks |
| 12:10 - 13:30 | Lunch |
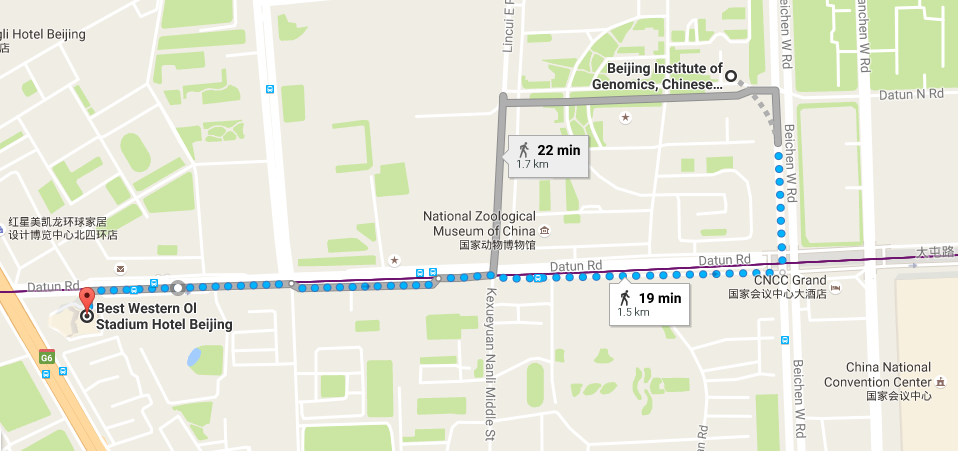
Location
-
Beijing Institute of Genomics, CAS
No.1 Beichen West Road, Chaoyang District
Beijing 100101, China -

Hotel
-
Best Western Ol Stadium Hotel (北京亚奥国际酒店)
No.1 Datun Road Jia, Chaoyang District
Beijing 100101, China -