The human microbiome, that is the totality of microbes inhabiting us, is spread over various body sites, the largest and probably medically most relevant one is the human gut. The gut microbiome is a complex system, which is still little understood and it is not even clear how the healthy state(s) should be defined. Yet, an increasing number of diseases is being associated with dysbiosis of the microbiome, often implied by metagenome-wide association studies (MWAS). MWAS do not reveal causalities, but disease associations and thus can still be a starting point for diagnostics, as I will illustrate using colorectal cancer (Wirbel et al., Nature Med., 2019). MWAS results are also often indirect or confounded (Schmidt et al, Cell 2018), the latter we could demonstrate for type 2 diabetes, where the intake of the drug metformin rather than the disease itself leads to the association with the gut microbiome (Forslund et al., Nature 2015). In an in vitro screen of 1200 marketed drugs against each of 40 human gut bacteria we could show that a quarter of all non-antibiotic drugs directly inhibit at least one gut microbial strain (Maier et al., Nature, 2018), a taxonomic resolution level that is becoming also achievable in MWAS (Schloissnig et al. 2013). Using single nucleotide variants (SNVs) we are able, for example, to trace donor and recipient SNVs after faecal microbiota transplantation, a microbial therapy (Li et al., Science 2016), or to trace oral-gut transmission (Schmidt et al, elife 2019). As our microbes are coming from the environment and our planet is one big ecosystem, it is crucial to study biodiversity and interactions of microbes at planetary scale. The feasibility of such a global approach is illustrated by (i) the TARA oceans project, surveying the microbial diversity of this vast ecosystem by studying plankton in all major ocean regions (Bork et al., Science 2015 and refs therein) and (ii) topsoil microbiomics (Bahram et al., Nature, 2018), revealing a global war between fungi and bacteria as well as regional antibiotic resistance gene reservoirs.
The 4th Big Data Forum for Life and Health Sciences
The 4th Big Data Forum for Life and Health Sciences (October 13-16, 2019)
Biological research has entered the era of big data, including a wide variety of omics data and covering a broad range of health data. Such big data is generated at ever-growing rates and distributed throughout the world with heterogeneous standards and diverse limited access capabilities. However, the promise to translate these big data into big knowledge can be realized only if they are publicly shared. Thus, providing open access to omics & health big data is essential for expedited translation of big data into big knowledge and is becoming increasingly vital in advancing scientific research and promoting human healthcare and precise medical treatment.
Open Biodiversity & Health Big Data
会议地址: 中国科学院北京基因组研究所一层报告厅
It is our great pleasure to announce that the 2019 Big Data Forum for Life and Health Sciences will be held in October 13-16, 2019. A few renowned biomedical data scientists have agreed to give speeches. Likely, you are also cordially invited to share your work and participate in this excited event.
Looking forward to seeing you in Beijing, China! We will be working hard to ensure your stay not only a fruitful one, but also an enjoyable one!
Organizing Committee
- Zhang Zhang (Chair, BIG, CAS)
- Yiming Bao (BIG, CAS)
- Wenming Zhao (BIG, CAS)
- Jingfa Xiao (BIG, CAS)
- Songnian Hu (BIG, CAS)
- Jun Yu (BIG, CAS)
- Jingchu Luo (Peking University)
Previous Conferences
- The 3rd Big Data Forum for Life and Health Sciences, 2018 (Invited speakers: Luonan Chen, Frank Eisenhaber, Michael Galperin,...)
- The 2nd Big Data Forum for Life and Health Sciences, 2017 (Invited speakers: Alex Bateman, Runsheng Chen, Chris Sander,...)
- The 1st Big Data Forum for Life and Health Sciences, 2016 (Invited speakers: Domenica D'Elia, Erik Bongcam-Rudloff, Jun Yu,...)
Invited Speakers

Peer Bork
Professor
Structural and Computational Biology Unit
EMBL
Germany

Janusz M. Bujnicki
Professor of Biological Sciences
Laboratory of Bioinformatics and Protein Engineering
International Institute of Molecular and Cell Biology in Warsaw
Poland

Weihua Chen
Professor
College of Life Science & Technology
Huazhong University of Science and Technology
China

Xiangjun Du
Professor
School of Public Health (Shenzhen)
Sun Yat-sen University
China

Lizhi Gao
Professor
Kunming Institute of Botany
Chinese Academy of Sciences
China

Dianjing Guo
Associate Professor
School of Life Sciences
The Chinese University of Hong Kong, China
China

Heui-Soo Kim
Dean
College of Natural Sciences
Pusan National University
Korea

Chwan-Chuen King
Professor
Epidemiology and Preventive Medicine
Taiwan University
China

Sofia Kossida
Professor
University of Montpellier
IMGT®, the international ImMunoGeneTics information system®
France

Shelan Liu
Professor
Zhejiang Provincial Center for Disease Control and Prevention
China

Yuwen Liu
Professor
Agricultural Genomics Institute
Chinese Academy of Agricultural Sciences
China

Jian Lu
Professor
Center for Bioinformatics
School of Life Sciences, Peking University
China

Lijia Ma
Professor
School of Life Sciences
Westlake University
China

Jiao Li
Professor
Institute of Medical Information and Library
Chinese Academy of Medical Sciences
China

Rasmus Nielsen
Professor
Department of Statistics
University of California, Berkeley
United States

Jeffrey Townsend
Professor
Center for Biomedical Data Science
Yale University
USA

Ana Tereza Ribeiro de Vasconcelos
Head of the Bioinformatics Laboratory
National Laboratory of Scientific Computation
Bionformatics Laboratory
Brazil

Chen Wu
Professor
Cancer Hospital
Chinese Academy of Medical Sciences
China

Wen Wang
Professor
Research Center For Ecology and Environmental Science
Northwestern Polytechnical University
China

Xingming Zhao
Professor
Institute of Science and Technology for Brain-Inspired
Fudan University
China

Zhaolei Zhang
Professor
Donnelly Centre for Cellular and Biomolecular Research
University of Toronto
Canada
Agenda
The conference features selected talks (15mins) and lightning talks (5mins). Please submit your abstract for
consideration of oral presentation, particularly for junior researchers/postdocs/graduate students.
Online registration and abstract submission: https://bigd.big.ac.cn/cms/
On-site registration: First Floor, BIG
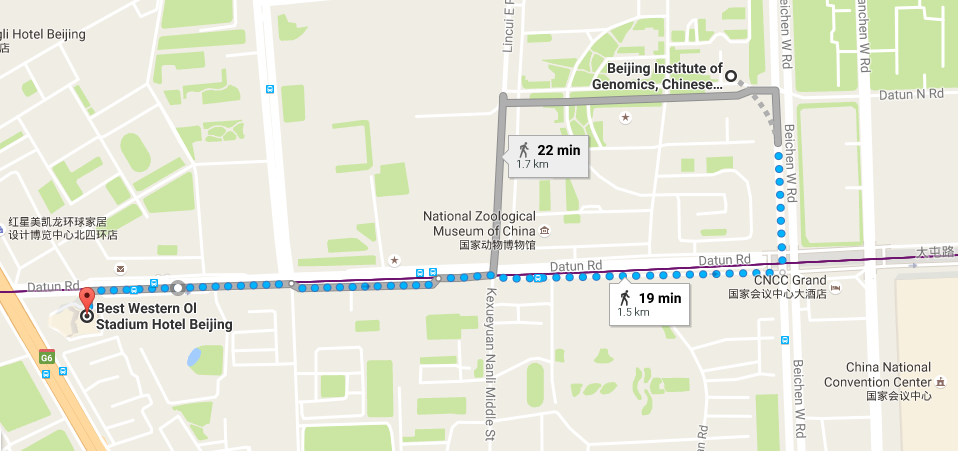
Conference Location(会议地址): Conference Hall at 1st floor, Beijing Institute of Genomics, Chinese Academy of Sciences, NO.1 Beichen West Road, Chaoyang District, Beijing 100101, China
| October 13 Sunday: Registration (First Floor, BIG) | |
|---|---|
| October 14 Monday: Talks (Conference Hall, First Floor, BIG) | |
| 09:00 - 10:35 | Session 1, chaired by Zhang Zhang, BIG, CAS |
| 09:00 - 09:25 | Welcome and Opening Remarks Zhang Zhang, On Behalf of the Organizing Committee Introduction of the Alliance of International Science Organizations Prof. Likun Ai, Assistant Executive Director of ANSO Secretariat |
| 09:25 - 10:05 | Microbiome analysis of the human gut and beyond Peer Bork, EMBL, Germany [Abstract]
|
| 10.05 - 10:35 | Single Cell Functional Genomics and Endometrium Cell Atlas Lijia Ma, Westlake University, China [Abstract]
The widely applied single cell genomic technology accelerates the researches on cell heterogeneity at the levels of DNA, gene expression profiles and chromatin topology by labeling individual cells with cellular barcode. We applied this technology in analyzing the endometrium tissue from Recurrent Implantation Failure (RIF) patients and healthy controls and characterized the distinct cell compositions between them. We demonstrated that the diversity of stromal cell of RIF patients is significantly lower than healthy control, which is consistent with their differences in endometrial lining thickness. We also detected a gene expression pattern that matches mid-to-late luteal phase in the RIF patients but not healthy control, which indicates an advanced or accelerated menstrual cycle in these patients. This study provides an insight of applying the single cell technology to decipher the underlying cell composition during endometrium cycle, and directly connected the lower cellular diversity and advanced gene expression profiles to clinic symptom. |
| 10:35 - 10:55 | Group Photo and Tea & Coffee Break |
| 10:55 - 12:25 | Session 2, chaired by Lijia Ma, Westlake University, China |
| 10:55 - 11:25 | Database Resources of the National Genomics Data Center Yiming Bao, BIG, CAS |
| 11:25 - 11:55 | Epigenetic feature in gastric cancer Dianjing Guo, The Chinese University of Hong Kong |
| 11:55 - 12:25 | Mining novel biomarkers from gut metagenomics data for patient stratification Weihua Chen, Huazhong University of Science and Technology, China |
| 12:25 - 14:00 | Lunch and BIG tour |
| 14:00 - 15:40 | Session 3, chaired by Yiming Bao, BIG, CAS |
| 14:00 - 14:40 | IMGT®, the international ImMunoGeneTics information system®: 30 years of immunoinformatics,
present endeavors and perspectives Sofia Kossida, University of Montpellier, France [Abstract]
IMGT®, the international ImMunoGeneTics information system® http://www.imgt.org,is the global reference in immunogenetics and immunoinformatics, created in 1989 by Marie-Paule Lefranc (Université de Montpellier and CNRS). IMGT® is a high -quality integrated knowledge resource specialized in the immunoglobulins (IG) or antibodies , T cell receptors (TR), major histocompatibility (MH) of human and other vertebrate species , and in the immunoglobulin superfamily (IgSF), MH superfamily (MhSF) and related proteins of the immune system (RPI) of vertebrates and invertebrates. IMGT® provides a common access to sequence, genome and structure Immunogenetics data, based on the concepts of IMGT-ONTOLOGY and on the IMGT Scientific chart rules. IMGT® works in close collaboration with EBI (Europe) , DDBJ (Japan) and NCBI (USA). IMGT® consists of sequence databases, genome database, structure database, and monoclonal antibodies database, Web resources and interactive tools. |
| 14:40 - 15:10 | Decoding brain with multi-dimensional data: An introduction of the Zhangjiang International
Brainbank Xingming Zhao, Fudan University, China |
| 15:10 - 15:40 | Annotation of human lncRNAs based on multi-omics data integration and analysis Lina Ma, BIG, CAS |
| 15:40 - 16:00 | Tea & Coffee Break |
| 16:00 - 17:55 | Session 4, chaired by Wenming Zhao, BIG, CAS |
| 16:00 - 16:40 | Large-scale ruminant genome data provides insighs into their evolution and distinct traits
Wen Wang, Northwestern Polytechnical University, China |
| 16:40 - 16:55 | Co-expressed gene-set enrichment analysis for drug repositioning with examples of psoriasis
and periodontal diseases Zhilong Jia, Chinese PLA General Hospital |
| 16:55 - 17:25 | Data-Driven Medical Studies: Efforts from National Scientific Data Center for Population and
Health Jiao Li, Institute of Medical Information and Library, Chinese Academy of Medical Sciences [Abstract]
Medical research paradigm is changing, from symptom-based medicine to evidence-based medicine to precision medicine. Nowadays, at the very beginning of a medical study, the principle investigator needs to make detailed data management plan (DMP), indicating the scientific data storage, data license, data sharing, data preservation and etc. In this talk, I will introduce the efforts made by the National Scientific Data Center for Population and Health on scientific data collection, data curation, data sharing, and data management tools development. Furthermore, I’d like to discuss the technical and management challenges in data-driven medical studies. |
| 17:25 - 17:55 | Less is more: Cancer cells evolve to use amino acids more economically
Jian Lu, Peking University, China [Abstract]
Rapidly proliferating cancer cells have much higher demand for proteinogenic amino acids than normal cells. The use of amino acids in human proteomes is largely affected by their bioavailability, which is constrained by the biosynthetic energy cost in the living organisms. Conceptually distinct from gene-based analyses, we introduce the energy cost per amino acid (ECPA) to quantitatively characterize the use of 20 amino acids during protein synthesis in human cells. By analyzing gene expression data from The Cancer Genome Atlas, we find that cancer cells evolve to utilize amino acids more economically by optimizing gene expression profiles. We further validate this pattern in an experimental evolution of xenograft tumors. ECPA not only shows robust prognostic power across many cancer types, but also improves the prediction of tumor response to checkpoint inhibitor immunotherapy. Our ECPA analysis reveals a common principle during cancer evolution. |
| 18:30 - 20:30 | Welcome Dinner |
| October 15 Tuesday: Talks (Conference Hall, First Floor, BIG) | |
| 13:00 - 14:35 | Session 5, chaired by Jingfa Xiao, BIG, CAS |
| 13:00 - 13:40 | Application of next generation sequencing in leukemia – from bench to bedside Zhaolei Zhang, University of Toronto, Canada [Abstract]
I will discuss our efforts in applying high-throughput DNA and RNA sequencing in the diagnostics and longitudinal monitoring of leukemias patients. We show that targeted sequencing is effective in ascertaining the mutation load after chemotherapy or allogeneic hematopoietic transplant, which in turn can serve as a good prognostic indicator. I will also discuss how such longitudinal mutation data can be used to trace the clonal evolution in leukemia patients. I will further discuss how patient’s electronic health data can be used in predicting the risk of developing leukemia. |
| 13:40 - 13:55 | Genomic Variant Information and Knowledge Databases
Shuhui Song, BIG, CAS |
| 13:55 - 14:35 | Genomic Impact of Transposable Elements and Its Biological Function Heui-Soo Kim, Pusan National University, Korea [Abstract]
Transposable elements can influence gene transcript ion and biological function through various mechanisms. Long terminal repeats (LTRs) of endogenous retroviruses (ERVs) have been shown to influence the expression of neighboring genes. Solitary LTRs contain various transcriptional regulatory elements including promoters, enhancers, and polyadenylation signals. They provide miRNAs during species radiation. Hypomethylation of the LTR element allows the neighboring functional gene to have tissue specific expression. Accumulated changes of the LTR elements in gene regulation are likely to be functional factors for the process of diversification, speciation and evolution consequences. A small minority of such sequences has acquired a role in regulating gene expression, and some of these may be related to differences between individuals, and to expression of disease. They seemed to be a source of alternative splicing, structural change of genomes, chromosome evolution, and could be related to genetic variation and epigenetic regulation linked to various diseases in various species. |
| 14:35 - 14:55 | Tea & Coffee Break |
| 14:55 - 16:35 | Session 6, chaired by Lina Ma, BIG, CAS |
| 14:55 - 15:35 | Structural bioinformatics of RNA molecules: integration of data from various sources at
different stages of computational 3D structure modeling Janusz Bujnicki, International Institute of Molecular and Cell Biology in Warsaw, Poland [Abstract]
Ribonucleic acid (RNA) molecules are master regulators of cells. They are involved in a variety of molecular processes: they transmit genetic information, sense cellular signals and communicate responses, and even catalyze chemical reactions. As in the case of proteins, RNA function is dictated by its structure and by its ability to adopt different conformations, which in turn is encoded in the sequence. Understanding how these molecules interact to carry out their biological roles requires detailed knowledge of RNA 3D structure and dynamics as well as thermodynamics, which strongly governs the folding of RNA and RNA-RNA interactions as well as a host of other interactions within the cellular environment. Experimental determination of these properties is difficult, and various computational methods have been developed to model the folding of RNA 3D structures and their interactions with other molecules. However, computational methods have limitations, especially when the biological effects demand computation of the dynamics beyond a few hundred nanoseconds. For the researcher confronted with such challenges, a more amenable approach is to resort to coarse-grained modeling to reduce the number of data points and computational demand to a more tractable size, while sacrificing as little critical information as possible. I will review strategies for computational modeling of RNA structure and dynamics, and for the structure determination of RNA interactions and complexes with other molecules (ions, small molecules, and proteins). I will show applications of SimRNA, a method developed in my laboratory, which uses a coarse-grained representation, relies on the Monte Carlo method for sampling the conformational space, and employs a statistical potential to approximate the energy and identify conformations that correspond to biologically relevant structures. I will also discuss the use of computational approaches for RNA structure prediction that can utilize data from experimental analyses. |
| 15:35 - 16:05 | Integrated Epidemiologic Information System for Dengue and Influenza - From Surveillance to Epidemiological Investigation and Public Health Policies Chwan-Chuen King, Taiwan University, China [Abstract]
Large-scale outbreaks of dengue and human influenza have frequently occurred in Taiwan during summer and winter seasons, respectively. Facing challenges of fast increasing numbers of cases and deaths, an integrated epidemiological information system which provides timely data for decision-makers is necessary. Since March 2004 [i.e. after the outbreak of severe acute respiratory syndrome (SARS) in 2003], we established a hospital emergency department-based syndromic surveillance system (ED-SSS) that involves 11 syndrome groups capable of automatic transmission of daily patient data directly from hospitals with emergency care throughout Taiwan to the Centers for Disease Control in Taiwan (Taiwan-CDC). In addition, school-based infectious disease syndromic surveillance system (SID-SSS) was developed from Taipei City government in response to the 2009 influenza A (H1N1) pandemic. Teachers and nurses from preschools to universities in all 12 districts of Taipei City are required to report cases of symptomatic children or sick leaves on a daily bases through the SID-SSS. The SID-SSS has extended to New Taipei City and other parts in Taiwan. In other words, both ED-SSS and SID-SSS can provide daily epidemi-ological information when outbreaks of emerging infectious diseases caused by novel agents [e.g. enterovirus 71, 2009 swine influenza virus (pdmH1N1/09)] occur. In 2015, the most severe and largest epidemic of dengue occurred in southern Taiwan. A risk factor-based integrated dengue information system (RF-IDIS) which involve important risk factors (e.g. mosquito indices, meteorological factors, land use, high risk locations etc.) to assist outbreak investigation and risk management. This RF-IDIS was firstly established in Pingtung in 2016 and subsequently extended to other cities/counties in Taiwan. Source reduction of Aedes mosquitoes can start early from entomological surveillance data even before the detection of human cases. In the future, integrated influenza surveillance systems need to have human vaccine history, viral sequences of the virus derived from different host species, and ecology. |
| 16:05 - 16:35 | Big Data Integration Based on Mechanistic Model Xiangjun Du, School of Public Health (Shenzhen), Sun Yat-sen University, China [Abstract]
Take transmission dynamics of seasonal influenza virus as an example, I will show you the idea of using theoretical modelling to address critical questions in life and health sciences by integrating big data from multiple sources. I will show you the successes of long-term incidence forecasting of seasonal influenza virus recent years for the United States, and the challenges due to the complexity of pathogen evolution. Data integration based on mechanistic model is a way forward for study the complex diseases in life and health sciences. |
| 16:35 - 16:55 | Tea & Coffee Break |
| 16:55 - 18:20 | Session 7, chaired by Xiangjun Du, School of Public Health (Shenzhen), Sun Yat-sen University |
| 16:55 - 17:35 | Multi-omics analysis of esophageal squamous cell carcinoma reveals alcohol drinking-related mutation
signature and genomic alterations mediating interactions in tumor ecosystem Chen Wu, Cancer Hospital Chinese Academy of Medical Sciences |
| 17:35 - 17:50 | Computational analysis and visualization tools for 3D genomic study Juntao Gao, Tsinghua University |
| 17:50 - 18:20 | Inheritance and reprogramming of epigenomic landscape Jiang Liu, Beijing Institute of Genomics, Chinese Academy of Sciences |
| October 16 Wednesday: Talks (Conference Hall, First Floor, BIG) | |
| 09:00 - 10:20 | Session 8, chaired by Zhang Zhang, BIG, CAS |
| 09:00 - 09:40 | Mutation, selection, and the somatic evolution of cancer Jeffrey Townsend, Yale University, United States |
| 09:40 - 10:10 | Developing Experimental and Computational Methods to Study the Genetic Basis of Complex
Diseases and Traits in the Big Data Era Yuwen Liu, Agricultural Genomics Institute, Chinese Academy of Agricultural Sciences [Abstract]
With the accumulation of genotype and phenotype data over an increasing big number of individuals, genome-wide association studies have identified many genetic variants associated with complex diseases and traits. However, we lack high-throughput experimental tools to understand the biological functions of these variants. We also lack computational methods to integrate multi-omics data, or differenttypes of “big biology data”, to explore the genetic basis of complex diseases and traits. In my talk, I will present two experimental methods and one computational method that we have built/extended in the past in the field of human genetics. I will also talk a little bit about my future work focusing on utilizing big data in animal genetics to accelerate livestock genetic improvement. |
| 10:10 - 10:20 | CGVD: A genomic variation database for Chinese populations Jingyao Zeng, BIG, CAS |
| 10:20 - 10:40 | Tea & Coffee Break |
| 10:40 - 12:20 | Session 9, chaired by Rujiao Li, BIG, CAS |
| 10:40 - 11:20 | From virus to human genome through bioinformatics Ana Tereza Ribeiro de Vasconcelos, National Laboratory of Scientific Computation, Brazil [Abstract]
Bringing together the expertise of bioinformaticians and geneticists is crucial, since very specific and fundamental computational approaches are required for virus, microorganism and human genome research, particularly in an era of big data. Improve existing analytical tools, computational resources, data sharing approaches, new diagnostic tools, and bioinformatic training are crucial. In this talk I will present results in collaboration with several research groups in Brazil related to the Zika virus, neglected and genetic diseases and resistance to antibiotics. |
| 11:20 - 11:50 | Effects of long-term exposure to air pollution on the resurgence of scarlet fever in China:
a 15-year surveillance study Shelan Liu, Zhejiang Provincial Center for Disease Control and Prevention, China [Abstract]
Background: Some researches have projected a
relationship between scarlet fever and meteorology and air pollution in a specific regions
or cities, but the resurge mechanism of scarlet fever after 2011 in China has not
interpreted in view of nationwide. |
| 11:50 - 12:20 | The tea tree genome provides insights into tea-processing suitability, tea flavor and independent evolution of caffeine biosynthesis. Lizhi Gao, Kunming Institute of Botany, Chinese Academy of Sciences |
| 12:20 - 13:30 | Lunch |
| 13:30 - 15:15 | Special Session: The CAS Youth Innovation Promotion Association chaired by Shuhui Song and Lili Hao, BIG, CAS |
| 13:30 - 14:15 | 3 Selected talks, 15min/talk:
|
| 14:15 - 15:15 | 12 Lightning talks, 5min/talk:
|
| 15:15 - 15:30 | Tea & Coffee Break |
| 15:30 - 16:50 | Session 10, chaired by Weiwei Zhai, Institute of Zoology, CAS |
| 15:30 - 15:45 | Cross-sectional whole-genome sequencing and epidemiological study of multidrug-resistant Mycobacterium tuberculosis in China Cuidan Li, BIG, CAS |
| 15:45 - 16:00 | Genome Warehouse: A Public Repository Housing Genome-scale Data Meili Chen, BIG, CAS |
| 16:00 - 16:40 | Human adaptation to local environments Rasmus Nielsen, UC Berkeley, United States |
| 16:40 - 16:50 | Closing Remarks Zhang Zhang, BIG, CAS |
Conference Location (会议地址)
-
BIG Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences
1 Beichen West Road, Chaoyang District, Beijing 100101
中国科学院北京基因组研究所,北京市朝阳区北辰西路一号院 -

Hotel
-
Best Western Ol Stadium Hotel (北京亚奥国际酒店)
No.1 Datun Road Jia, Chaoyang District
Beijing 100101, China -