

Cytosine methylation is one of the most important RNA epigenetic modifications. With the development of experimental technology, scientists attach more importance to RNA cytosine methylation and find bisulfite sequencing is an effective experimental method for RNA cytosine methylation study. However, rarely tools can directly deal with RNA bisulfite sequencing data efficiently at present. Hence, we have developed a specialized tool BS-RNA. Its annotation result is in BED (.bed) format, including locations, sequence context types (CG/CHG/CHH, H = A, T, or C), reference sequencing depths, cytosine sequencing depths, and methylation levels of covered cytosine sites on both Watson and Crick strands. BS-RNA supports both paired-end and single-end sequencing short reads from a directional bisulfite library. Evaluation results of the rates of uniquely mapped reads, rates of correctly mapped reads, and running time for simulated data and actual data indicate that BS-RNA can provide fast and accurate mapping of RNA bisulfite sequencing reads. Comparison between annotated cytosine methylation used BS-RNA and published research also shows that BS-RNA is an effective annotation tool for RNA bisulfite sequencing data.
The BS-RNA process (Figure. 1-2) includes three main steps: pre-treatment, mapping, and annotation.
The first step is the pre-treatment of reference genome sequences, sequencing data, and gene annotation file: (1) the reference genome sequence is converted twice in parallel as follows: (A) cytosines are replaced by thymines and (B) guanines are replaced by adenines. This genome sequence conversion only need to be performed the first time one uses the reference genome sequence, which means it could be reused for all the following analysis which use the same reference genome sequence. (2) cytosines in reads of the T-rich sequencing read file are replaced by thymines, while guanines in reads of the A-rich sequencing read file are replaced by adenines. And (3) the gene annotation file in GTF format is revised to fit the converted reference genome sequences. Each annotation line is converted twice simultaneously: "C-T" and "G-A" are appended to the chromosome label in the gene annotation file, respectively.
Next, the HISAT2 program is invoked by BS-RNA to build alternative splicing according to the modified annotation gene file and align pre-processed reads to the converted reference genome sequence. BS-RNA filters out two types of reads that are mapped to the reference genome sequence as follows: (1) reads mapped to multiple positions and (2) reads mapped to the wrong strands (T-rich reads mapped to reverse-complement of reference sequence converted C to T or to reference sequence converted G to A, A-rich reads mapped to reference sequence converted C to T or to reverse complements of reference sequence converted G to A). The original mapping file (SAM format), filtered mapping file (SAM format), and mapping report file will be provided by BS-RNA when the mapping step is finished.
Further, BS-RNA splits the filtered mapping file into two files: a Watson-strand SAM format file (mapped to reference converted C to T) and a Crick-strand SAM format file (mapped to reference converted G to A). SAMtools is embedded into BS-RNA to convert the Watson-strand and Crick-strand SAM format files to BAM format and then sort these two BAM files according to the chromosome coordination. Single-base coverage information is extracted using the mpileup command of SAMtools from the sorted BAM files. For each cytosine position in the genome, the read base (SAMtools mpileup output information) is deemed to support methylated cytosine sequencing if it matches a dot or a comma, otherwise it is deemed to support an unmethylated cytosine sequencing. We excluded the reference skips (which menas "<" or ">" is not counted). BS-RNA provides the annotation result for each covered cytosine in a BED (.bed) file with information related to the methylation character: location of the covered cytosine in the reference, sequence context type (CG/CHG/CHH, H = A, T, or C), reference sequencing depth (total number of reads mapped to the cytosine site), cytosine sequencing depth (total number of reads that supported a methylated cytosine at this site), and methylation level (the ratio of cytosine sequencing depth to sequencing depth at the cytosine site) on the Watson strand and Crick strands for each chromosome to the users. Annotation files in the BED format make it easy to observe the methylation distributions using an IGV or UCSC browser.
Documentation and usage information can be found here.
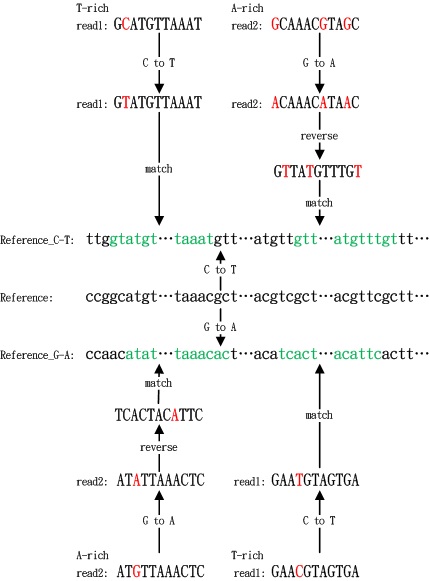
Fig.1. mapping principle of BS-RNA
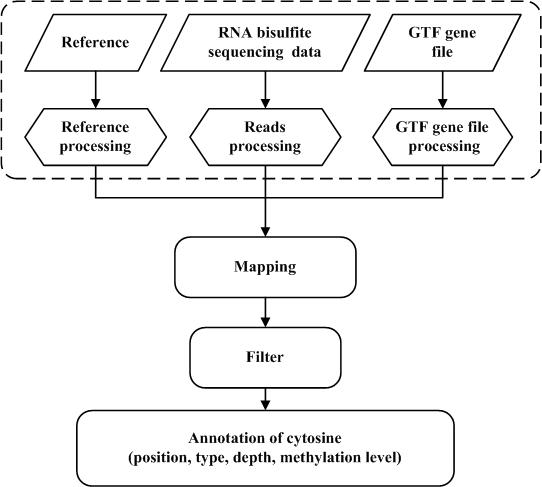
Fig.2. Flowchart of mapping and annotation of BS-RNA
To test the performance of BS-RNA, test sample data is provided here. The test data (simulated RNA bisulfite sequencing data set of human) is a paired-end dataset in FastQ format (Phred33). The length of read is 100bp. The command for testing the demo data could be like this:
BS-RNA_v1.0 --perlDir script --reads1 test_T-rich.fq --reads2 test_A-rich.fq --gene Homo_sapiens.GRCh37.75.gtf
--rawRef hg19_ref --pathToPython /.../python2.7.8/bin --pathToHISAT2 /.../hisat2-2.0.1-beta
--pathToSAMtools /.../samtools-0.1.16 --outDir /.../demo_resultIt will take about 2.2 hours to complete all the analysis for this test data (including indexing the reference genome sequences) on a standalone, 2.4 GHz single-core processor with 13G memory.
Please send bugs or advice to the author(lirj@big.ac.cn or liangf@big.ac.cn),thank you.