KaKs_Calculator adopts model selection and model averaging to calculate nonsynonymous (Ka) and synonymous (Ks) substitution rates, attempting to include as many features as needed for accurately capturing evolutionary information in protein-coding sequences. In addition, several existing methods for calculating Ka and Ks are also incorporated into KaKs_Calculator.
Documentation and help information can be found here.
The KaKs_Calculator package, including source codes, compiled executables and documentation, is freely available for academic use only at here.
The input sequences are AXT-formatted and please find information about how to generate multiple AXT sequences using ParaAT, a parallel tool capable for constructing a large number of protein-coding DNA alignments.
Please take a look at below example.
NP_000026
ATGCTCCTGTG-CCACTGGCC
ATCCCC-TGCGCTCACTGGAC
NP_000053
ACAGaTtCTACCc-GCCcACTA--GgtGtt
---ggTTCTCCtACCcA-G-CACTACTggg
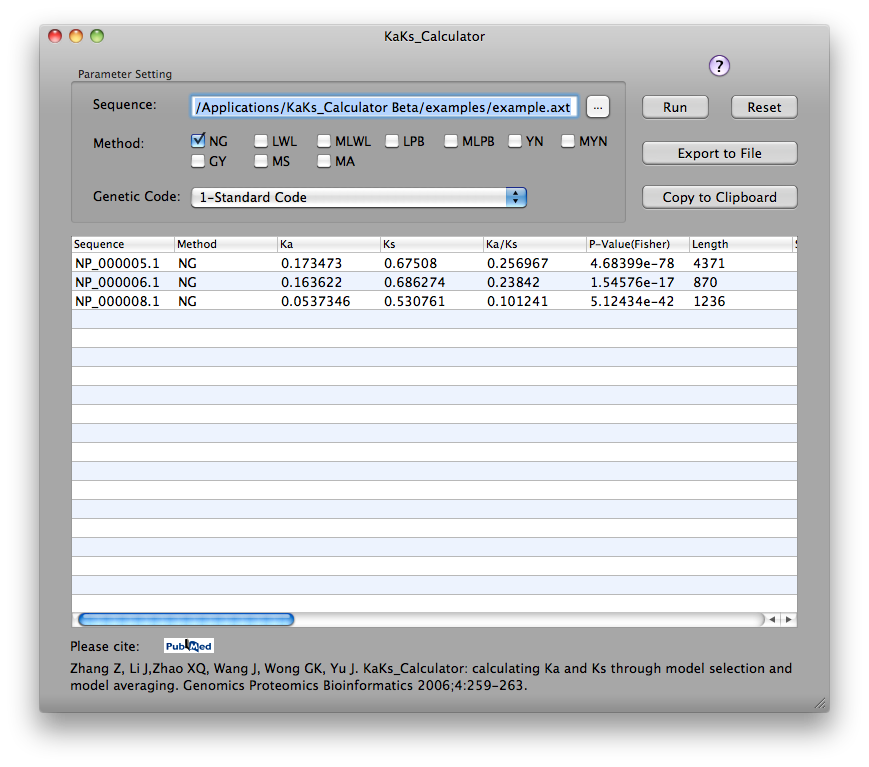
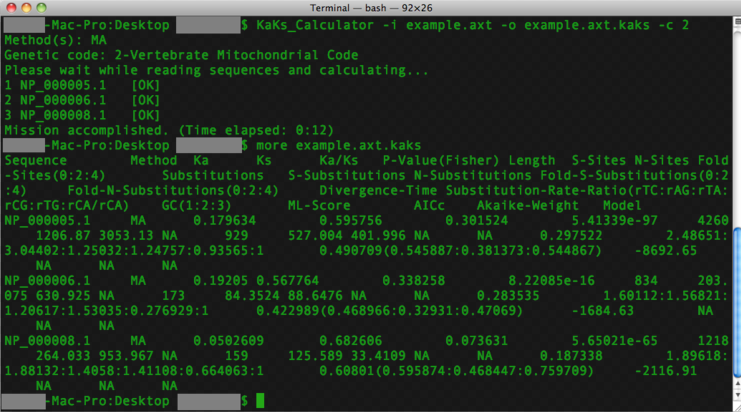
Screenshots on Windows with GUI, MAC with GUI and command line.
The answers to Frequently Asked Questions are summarized, e.g., regarding MYA (Million Years Ago) deduction from Ks, fisher exact test for nonsynonymous and synonymous substitutions and sites.
KaKs_Calculator has been used by a number of papers.
Reference:
Zhang Z, Li J, Zhao XQ, Wang J, Wong GK, Yu J: KaKs Calculator: Calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinformatics 2006 , 4:259-263.
How to cite:
For example: Nonsynonymous and synonymous substitution rates (denoted as Ka and Ks, respectively) were estimated using the software KaKs_Calculator (Zhang et al. 2006), with the method of model averaging (YN, MYN, GY, etc.).