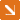 1. Search Process
1. Search Process
Delta.EPI is designed as a meta-tool to privide a click-and-go solution for EPI searching and visualization.
Briefly, researches with Hi-C experiment are curated and all Hi-C data are processed with the same standard workflow. In a single research, contact pairs from samples belonging to the same cell line and cultured under the same experimental conditions will be pooled together
to allow downstream high-resolution interaction analysis. This pooling process is done as is mentioned in the original research paper or additional materials. The pooled samples, sharing the same cell line and condition info, will be set as one entry in our database.
For each entry, interactions are called by five independent Hi-C data analysing softwares in advance. Users can set as input a locus, a piece of DNA sequences, or a gene identifier. Then, input information will be located on the genome using built-in tools as input span(s) and any interactions that have one bin overlapped with it will be selected. For those interaction pairs, any TSS regions that overlap with the other bin will be recorded, and such input-TSS pairs will be listed on the result page as putative interactions. By default, the interactions are sorted in the order of (1) amount of methods agree on this interaction (2) overlapped ratio of the bin and corresponding span, which means interactions with higher overall reliability should rank at the top.
Currently there are three search modes users may use to search for interactions. You could switch the search mode by simply clicking on three corresponding options in the Custom Search page.
-
Search for Locus
If users know the exact position of the enhancer( or other elements of interest), Locus Search mode could simply be used for analysis. Please make sure the locus uses the same reference genome as delta.EPI, and if they don't, users can easily convert them using UCSC online LiftOver tool.

-
Search for Sequence
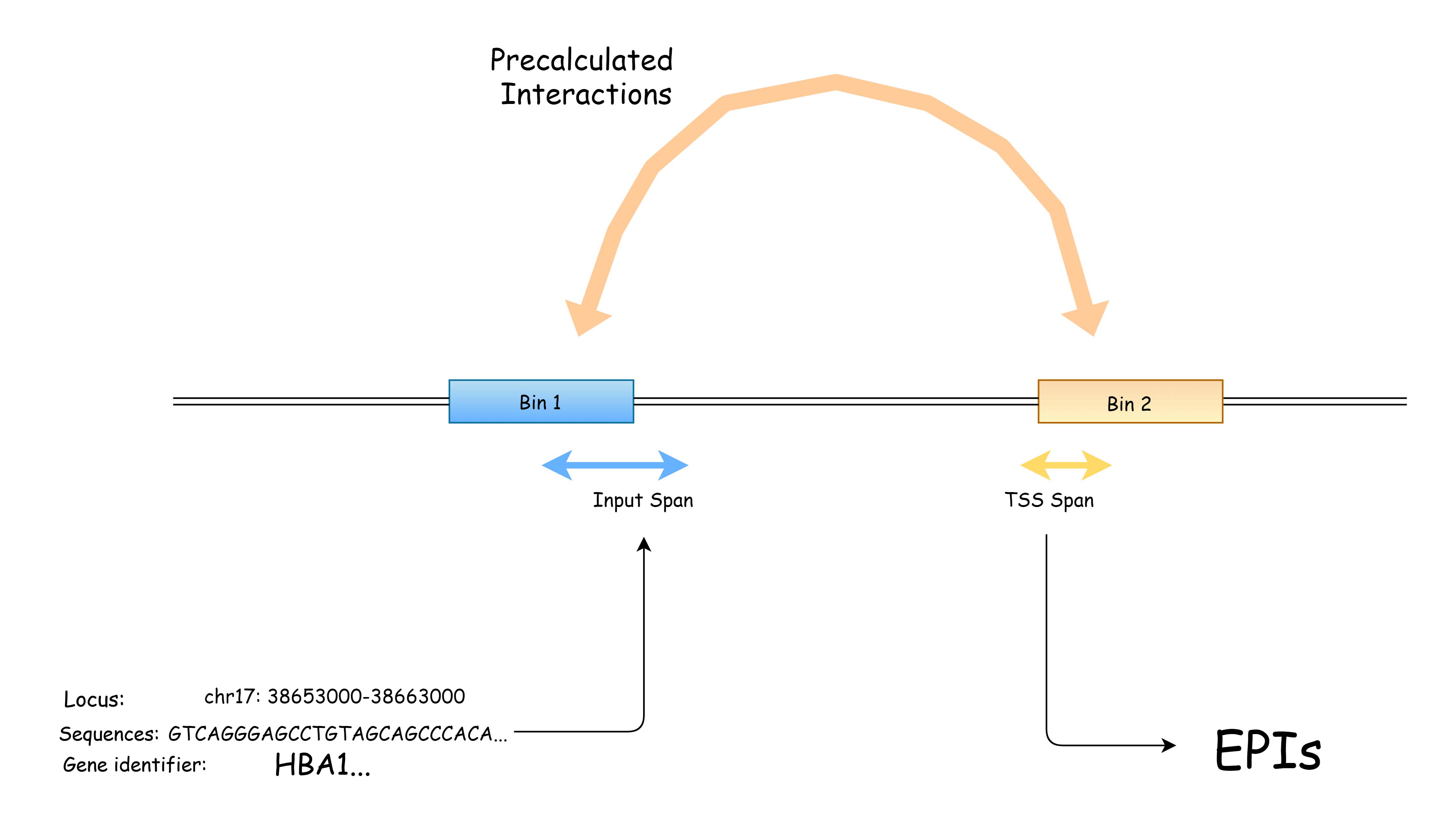
Sequence Search mode integrates an NCBI Blast Tools to provide support for users interesting in a specific DNA sequence while don't know where it come from. Inputted sequence will be mapped to the reference genome of selected species, and the result locus will be use as input for downstream processing. An editable span length variable decides the length of input span(extends from the midpoint of mapped sequence to upstream and downstream with same length).
Note that only the match with highest mapping score will be used, so if the sequence is too short or does not result in a considerably small q value, we highly recommand you use NCBI online Blast tool, mapping to reference genome and select the locus yourself.
-
Search for Gene Identifier
Users could also set gene identifier as search target. Note that in this mode, TSS(es) of the input gene will be set as input spans, so basically results of TSS-TSS interactions instead of EPIs will be generated. Supported gene identifiers include Gene Symbol(Alias), or accession numbers from frequently-used database such as GenBank, Unigene, Entrez, Ensembl, UniProt, Pfam, Prosite. For some sorts of identifiers, one-to-many search result may be given, in this case the user may distinguish them by the unique Entrez ID and select one or more of them.
The gene identifier info is extracted from OrgDb , and the TSS info is obtained from TxDb via R Bioconductor.

-
Quick Search
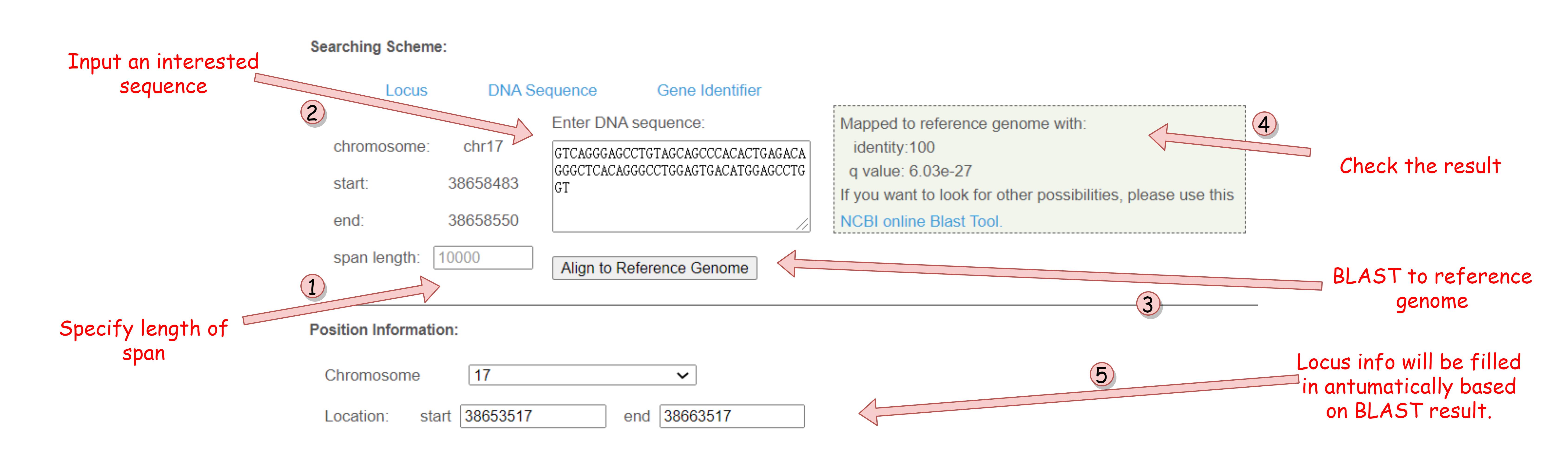
The Quick Search bar on the index page is a combination of three above search modes. By filling this single text box with some specific formats, users could arose corresponding search modes and get instant feedback.
- For Locus Search, users may need to use the format "chrN : start-end", where 'N','start','end' denotes the detailed info of the locus.
- For Sequence Search, users need to add a "seq:" tag before the whole sequence.
- All inputs that do not match to previous formats will be used in Identifier Search.
*: All formats used in quick search are space and case insensitive, so users may only need to make sure the tag(in black color) and search targets(in red color) are in proper order. you may also click on the examples below the quick search textbox to try it out.
*: Note that since it is not guaranteed that users' input will successfully map to the genome or match one identifier in the database, for Sequence or Identifier search mode, you may still need to click on Submit button to continue analysis after manually check the search result.
In the Custom Search Page, users could also select the cell lines info to narrow down the searching scope, the four selection bars are in
hierarchical structure, users may stop at any layers to get entries of one specific species, tissues, cell types or cell lines.
Users may also add some additional screening conditions such as only selecting result from control group to filter those results obtained under some special experimental condition. By default all entries that meet the cell line requirements will be listed in the Entry list on Result Page.