circAtlas 3.0 a gateway to 3 million standardized named vertebrate circular RNAs
How to use
Introduction
Welcome to circAtlas 3.0 (a gateway to consistently curated vertebrate circular RNAs).
circAtlas 3.0 contains a compendium of 2,674 RNA-seq libraries from 33 normal tissues from human, macaque, mouse, rat, rabbit, cat, dog, pig, sheep and chicken.
Main characteristics of circAtlas include:
circAtlas uses the standardized nomenclature scheme proposed by the circRNA community.
circAtlas integrates Illumina and Nanopore circRNA sequencing datasets, providing the full-length sequences and downstream functional analysis results of over 2.5 million circRNAs.
Convert the circRNA ID among different circRNA databases.
Search and find conserved circRNAs across human, macaque, mouse, rat, rabbit, cat, dog, pig, sheep and chicken.
Predict second structure of circRNAs.
Predict potential peptide-encoding circRNAs. e.g: ORF, IRES.
Predict regulation elements of circRNAs. e.g: miRNA, RBP.
circAtlas uses three types of networks including gene co-expression, miRNA regulation and RBP regulation to infer circRNA functions.
Dataset
circAtlas 3.0 comprises a comprehensive compendium of 2,609 Illumina and 65 nanopore RNA-seq datasets from 33 diverse tissues within 10 distinct species. More details are show in index page.
Browse circAtlas
Users can select one of the ten species to search for circRNAs.

Users can get the detailed information of each circRNA by clicking the circAltas ID.

Search circAtlas
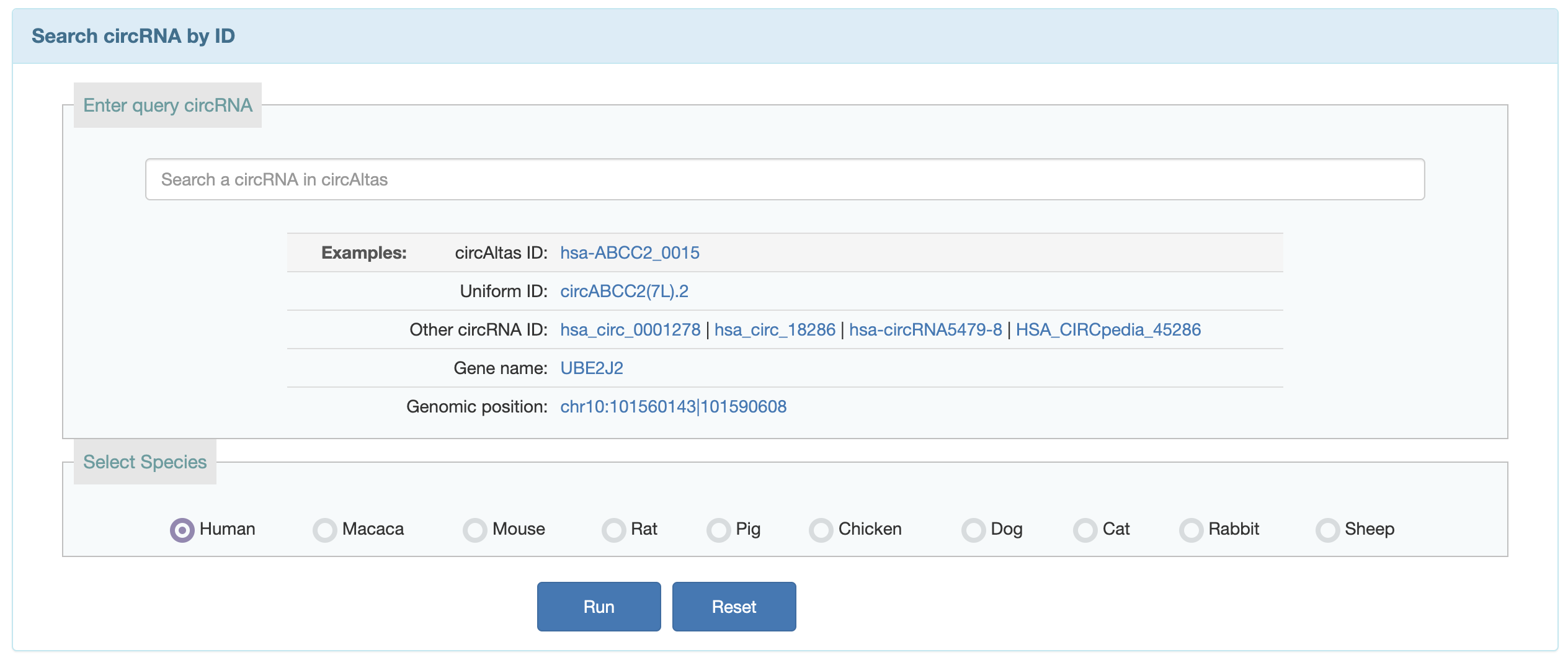
1. Users can search the circRNA known in circAtlas by circAtlas ID, chromosome position, host gene and name in other circRNA databases.
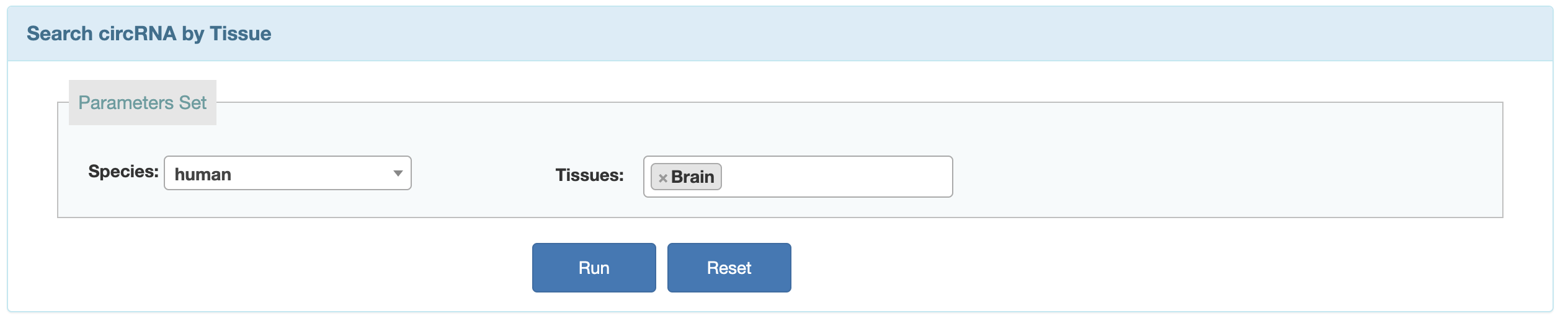
2. Users can search the circRNA by expressed species and tissues.
3. Users can search for the circRNAs that are known in the circAtlas and have a potential function, such as:
- Conversed circRNAs
- ORF
- IRES
- RBP
- miRNA
- Network annotation

circRNA names
circAltas provides two types of names for each circRNA:
circRNA sequences and isoforms
For Illumina RNA-seq data, the cleaned reads were aligned to the reference genome using bwa (v0.7.17), and circRNAs were subsequently identified and quantified using the CIRI2 (v2.0.6) and CIRIquant (v1.1.2) pipeline. To reconstruct the full-length sequence of circRNA isoforms, CIRI-full (v2.1.1) and CIRI-vis (v1.4.1) were employed to assemble full-length circRNAs using the reverse overlap feature. To further reduce the number of false positives , three other software, CIRCexplorer2 (v2.3.8), DCC (v0.5.0) and find_circ (v1.2), were also used to identify circRNAs, and circRNAs that are supported by at least one of the other tools were retained for further analysis.
For long-read sequencing data, the raw sequencing data from the CIRI-long protocol were trimmed using Porechop, and CIRI-long (v1.1.0) was performed to identify circRNAs from nanopore sequencing reads. For other long-read circRNA sequencing protocols including isoCirc, circFL-seq and an adapted version of CIRI-long, the processed data were obtained from their original manuscripts. Finally, the reconstructed full-length sequences were used to annotate the spliced isoforms of the identified circRNAs.
circRNA second structures
Based on the assembled full-length circRNA sequences, the secondary structure was predicted using the RNAfold algorithm of the Vienna RNA 2.0 package.
circRNA expressions
circAltas provides the expression of circRNAs across normal and cancer tissues(human only). circAltas integrates the expression landscape of human circRNAs in clinical samples across 36 cancer types from the MiOncoCirc and CSCD2 databases.
Analyze
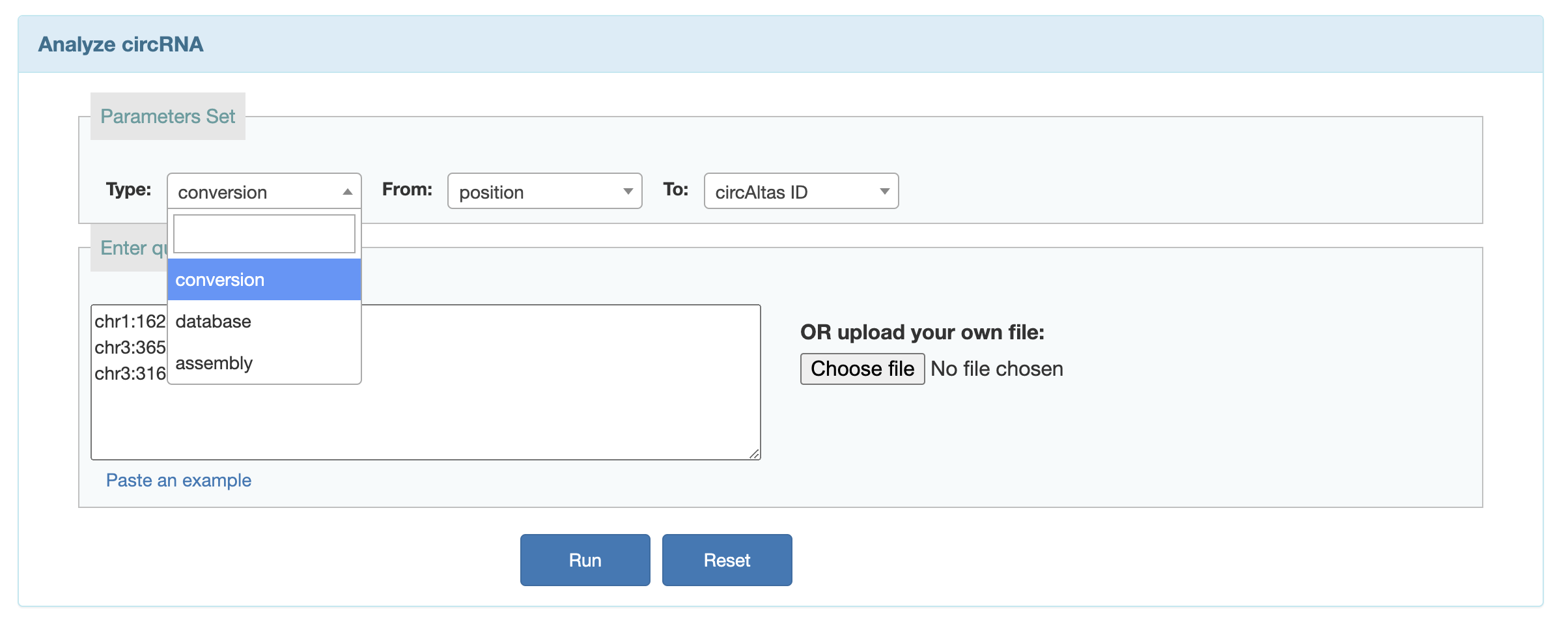
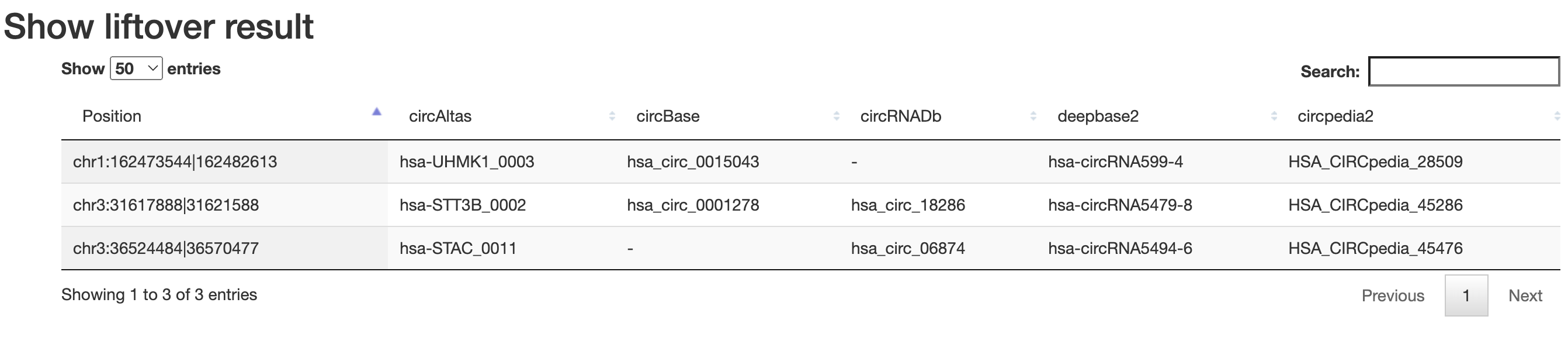
Convert(liftover)
Users can search for circRNAs in different databases and liftover circRNA position from different assemblies.
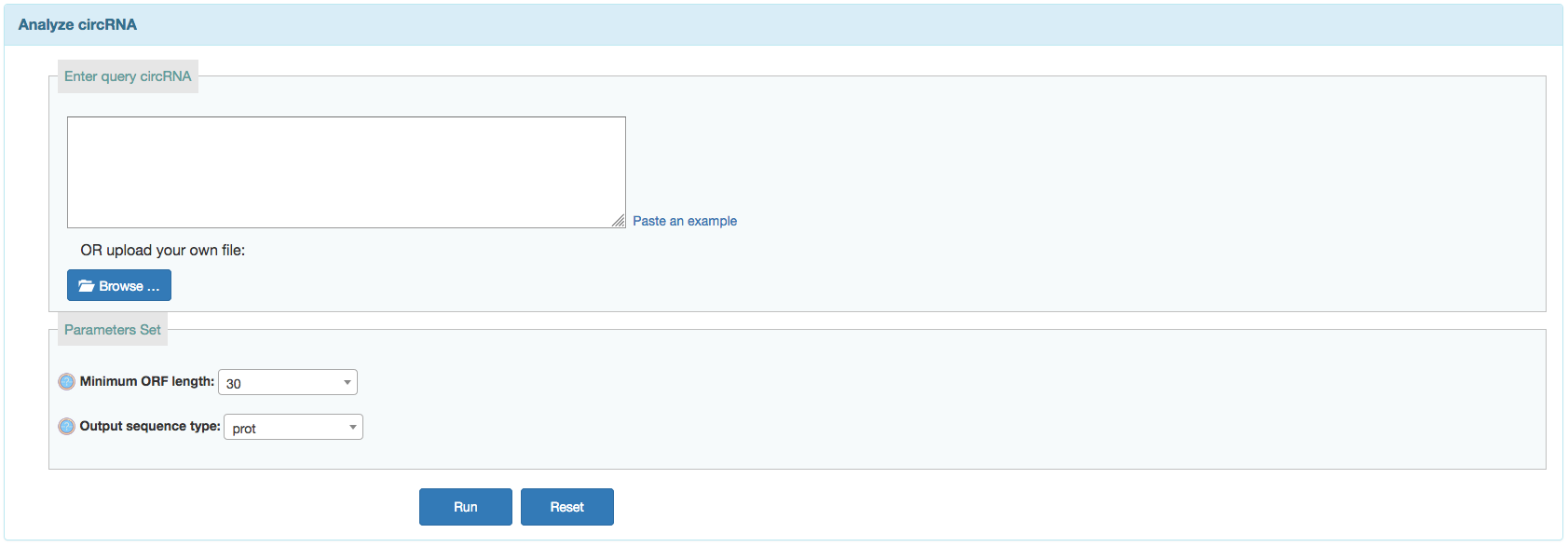
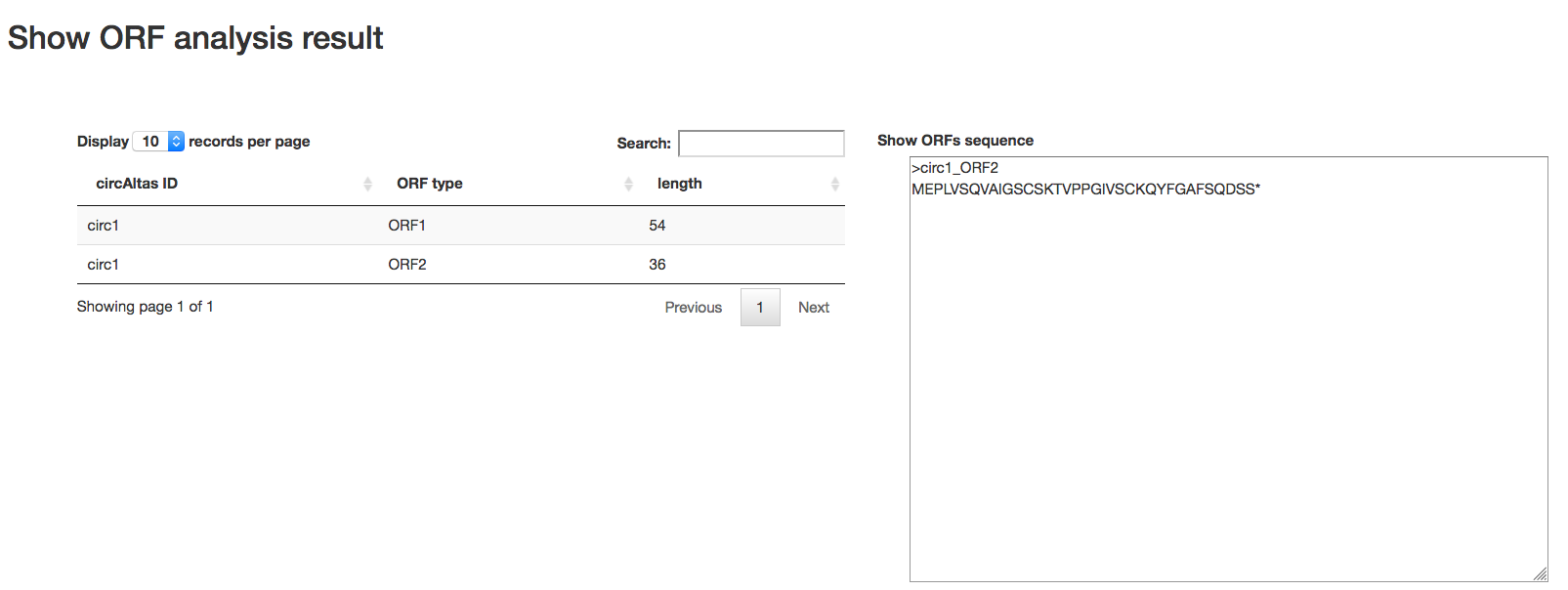
ORF
Users can search predicted IREs on currently available circRNAs or predict IREs on given circRNAs from a corresponding input.
- Min ORF length: Sets minimum sequence identity (in percent). Default is 60 for circRNAs searches.
- Output type: Sets minimum score. This is the matches minus the mismatches minus some sort of gap penalty. Default is 60.
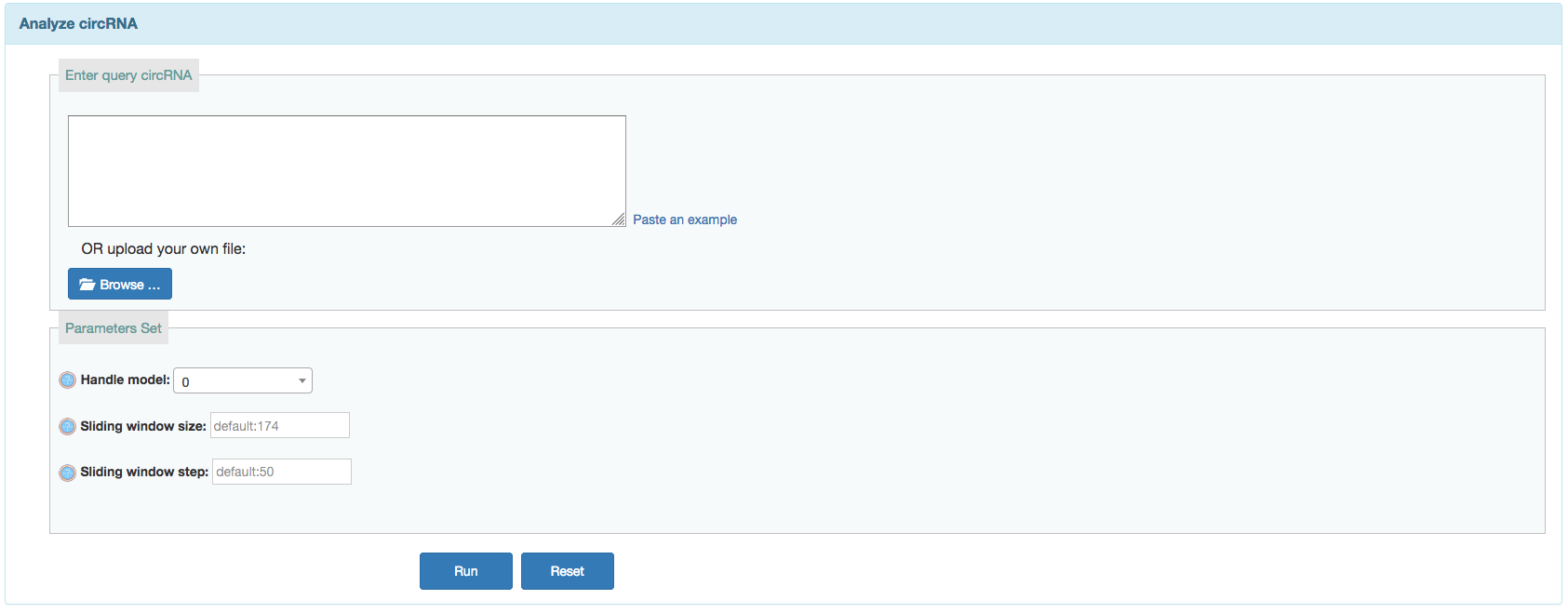
IRES
User can search predicted IREs on currently available circRNAs or predict IREs on given circRNAs from a corresponding input.
- Handel Model:
- 0,default, just using the complete transcript sequences;
- 1, optional, using the subsequences specified by the index (e.g. >id start_pos end_pos);
- 2, optional, spliting the transcript into fragments, please provides the window size (-w, defualt 174) and the step size (-s, defualt 50);
- Sliding window size: Ignore this option if the selected model is not 2. Default is 174.
- Sliding window step: Ignore this option if the selected model is not 2. Default is 50.

RBP
User can search predicted RBP binding on currently available circRNAs or predict RBP binding sites on given circRNAs from corresponding input.
- Region: Predict RBP bingding sites in upstream, downstream, and sequence.
- RNA-binding Proteins: The RNA-binding Protein all from POSTAR2 and starBase databases.Users can choose all factor or select several factors.
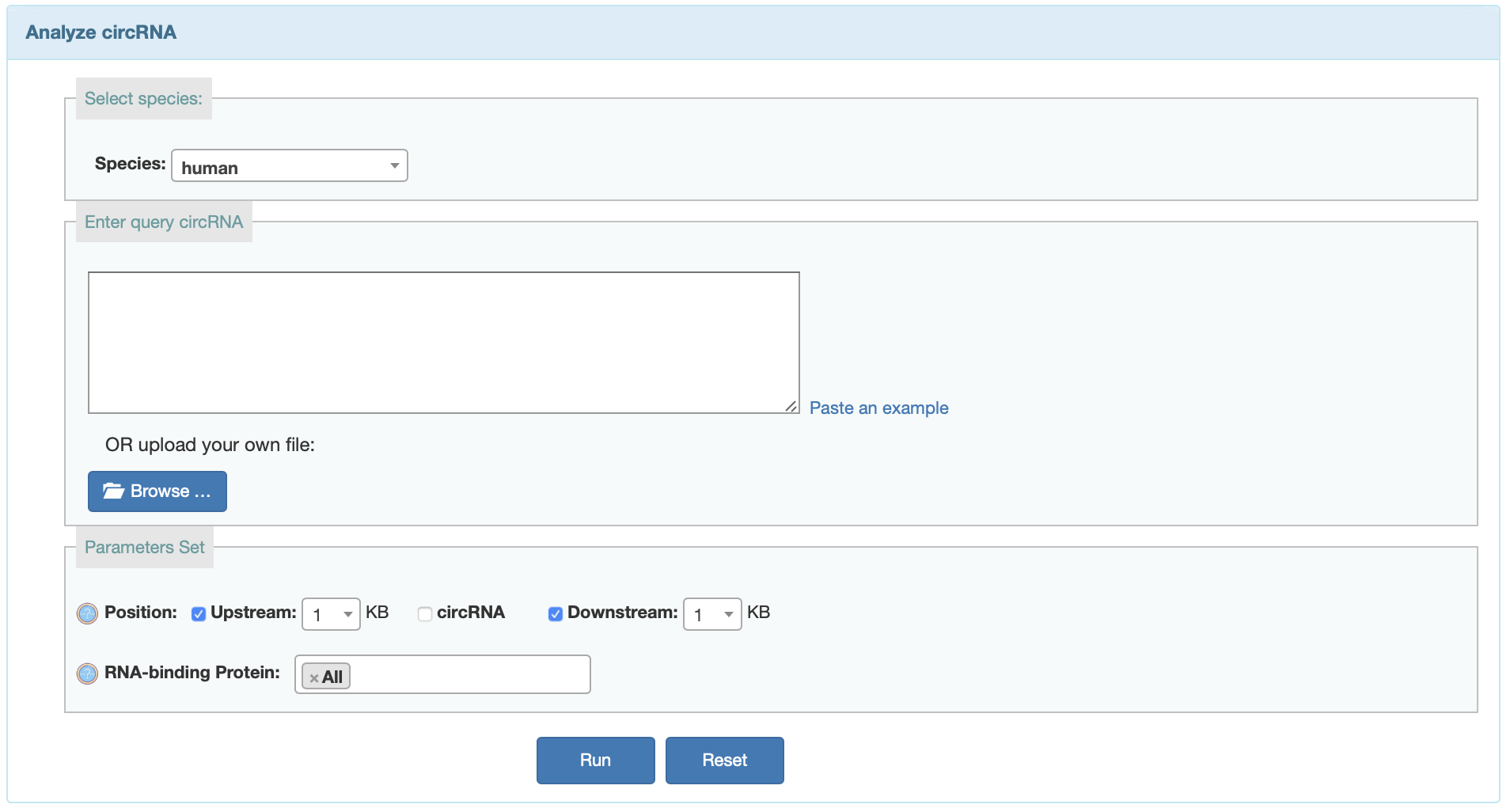
miRNA
User can search predicted miRNA binding on currently available circRNAs or predict miRNA binding sites on given circRNAs from acorresponding input.
- Software:Users can choose different software to predict the miRNA binding sites. Pita will take several minutes.

Network
User can search functional annotation of currently available circRNAs or annotate given circRNAs by constructing network.
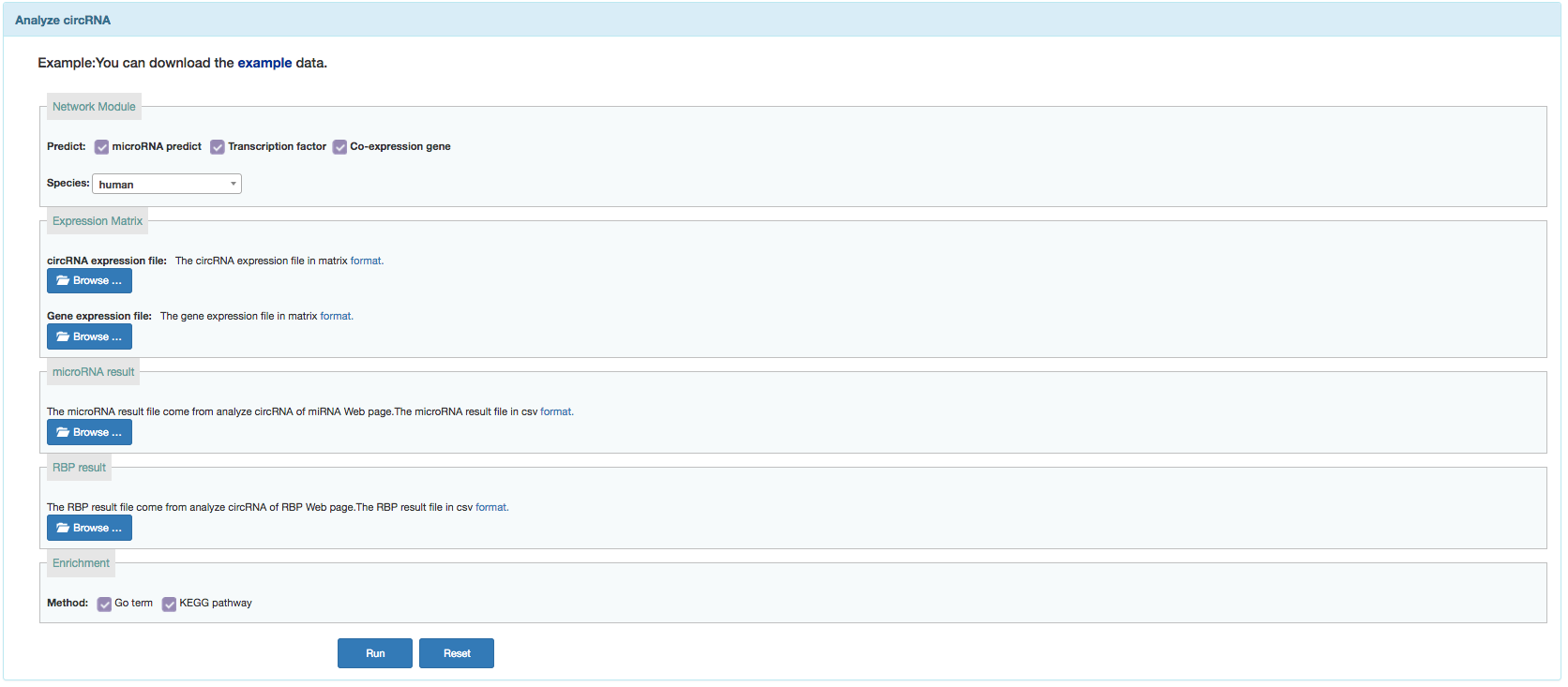
- circRNA expression matrix:in matrix format.
- gene expression matrix:in matrix format.
- miRNA result:in csv format.Download the result from miRNA result page.
- RPB result:in csv format.Download the result from RBP result page.
- Annotation method:GO term and KEGG pathway.
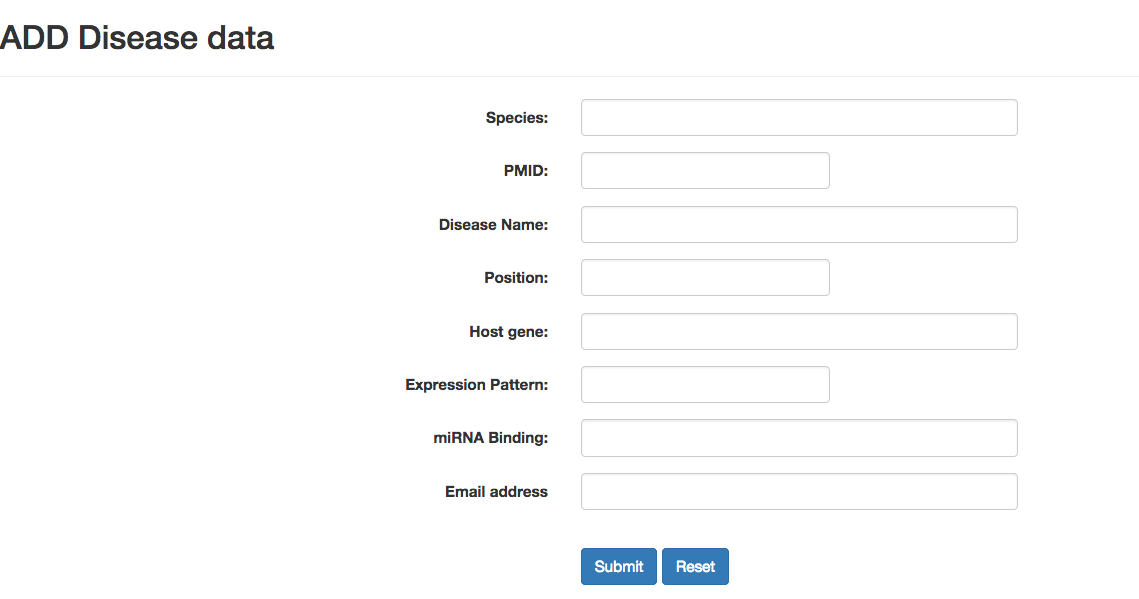
Disease
The page provides disease related circRNA by integrating three databases: circad, CircR2Disease and circRNADisease. Users can search and query target circRNAs using this module. Users can also submit new records by providing your work email address, where you will receive the confirmation request. If there are any problems with your future submissions, the circAtlas curators will contact you at this email address. Please note that your email address will be presented on the web in a way that should protect it from spammers.