Gene Expression
Nebulas
GEN分析工具包提供覆盖组织(bulk)和单细胞(10X Genomics,Smart-seq2,Drop-seq以及inDrop类型)转录组测序数据的一站式分析流程。GEN数据库中所有的基因/转录本表达谱都是基于GENToolkit统一分析处理的。GENToolkit由两个主要部分组成,分别是上游和下游分析流程。具体来说,上游分析模块包括4个步骤: “索引构建”,“质量控制”,“序列比对”,“基因表达定量”,同时下游分析模块包含2个重要步骤: “基因表达谱分析” 和 “分析结果可视化”。GENToolkit支持“sra”或“fastq”(单端或双端)格式的原始数据,用于进一步的基因/转录本表达谱分析。根据用户的需要,可以对数据集中的全部或部分样本进行基因表达分析。
tar -zvxf GENtoolkit.tar.gz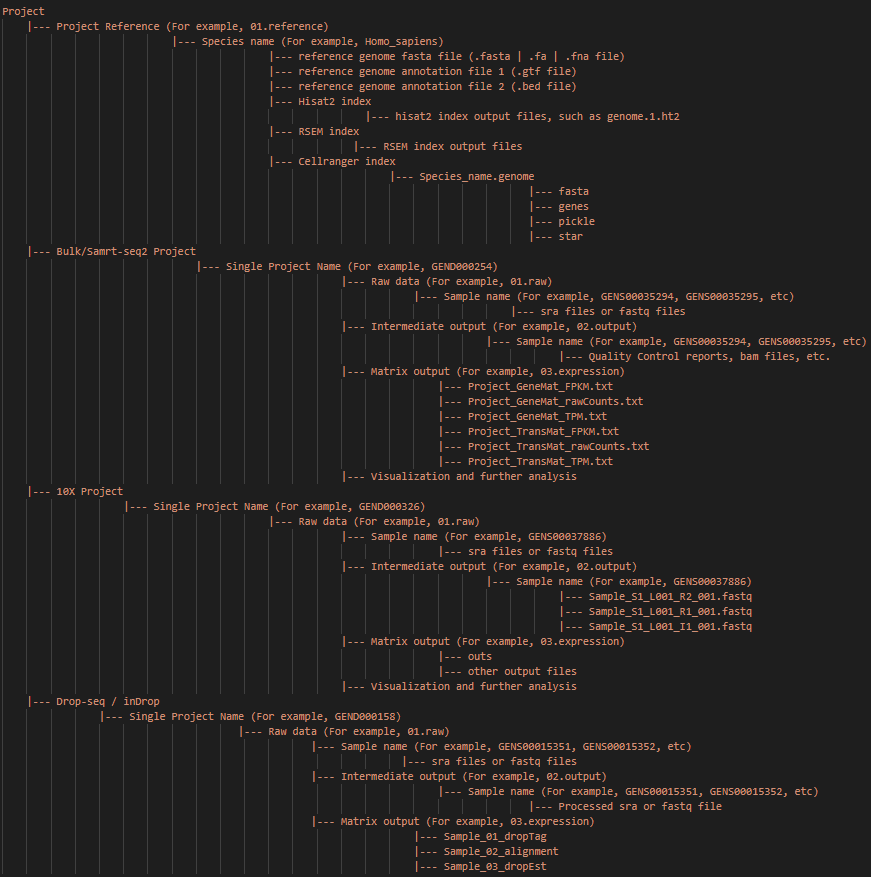
注: 整个项目的主要输入输出文件和路径如上图所示。推荐使用这种目录结构。
python GENtoolkit.py [options] ...python GENtoolkit.py [options] -blt Bulk -ipp
../Oryza_sativa -rgf reference.fasta -rgg reference.gtf -sp
../STAR/bin/Linux_x86_64 -rd ../01.raw -st pair -hi
../Oryza_sativa_hisat2/genome -ri
../Oryza_sativa_RSEM/Oryza_sativa -bf reference.bed
注: 测试数据来自于GEN (GEND000254 和 GEND000257)
python GENtoolkit.py [options] -blt Smart-seq2 -ipp ../Oryza_sativa -rgf reference.fasta -rgg reference.gtf -sp ../STAR/bin/Linux_x86_64 -rd ../01.raw -st pair -hi ../Oryza_sativa_hisat2/genome -ri ../Oryza_sativa_RSEM/Oryza_sativa -bf reference.bed
注: 测试数据来自于GEN (GEND000305)
python GENtoolkit.py [options] -blt 10X -ipp ../Saccharomyces_cerevisiae -rgf reference.fasta -rgg reference.gtf -sp ../STAR/bin/Linux_x86_64 -rd ../01.raw -ci ../Saccharomyces_cerevisiae/Saccharomyces_cerevisiae.genome
注: 测试数据来自于GEN (GEND000326)。如果由于内核、内存或其他原因的限制导致某些样本任务运行失败,可以通过添加"-da Designated_samples -sl sample1-sample2-sample3" 参数帮助任务程序重新运行。
python GENtoolkit.py -blt Drop-seq -ipp ../Homo_sapiens -rgf reference.fasta -rgg reference.gtf -si ../STAR/bin/Linux_x86_64 -dc reference.xml -dm ../mit_genes_human.ensembl.rds -rd ../01.raw
注: 测试数据来自于GEN (GEND000158). 如果已选择"-da Designated_samples"参数,则相应的"-sl []"参数必须添加!
python GENtoolkit.py [options] --stream down --BuildLibraryType Bulk --workpath ./workpath/ --exprData exprData.txt --metaData metaData.txt
python GENtoolkit.py [options] --stream down --BuildLibraryType Smart-seq2 --workpath ./workpath/ --exprData exprData.txt --metaData metaData.txt -- refpath ./ref/
python GENtoolkit.py [options] --stream down --BuildLibraryType 10X --workpath ./workpath/ --exprData ./data/ (including barcodes.tsv.gz, features.tsv.gz, matrix.mtx.gz) -- refpath ./ref/
| 长参数 | 短参数 | 描述 |
|---|---|---|
| --stream | -stream | Pipeline type, [ up | down | all ] |
| --IndexBuild | -ib | Once a reference genome is built, it can be used many times in a species gene expression analysis, which depends on whether building index or not. [ index_build | index_exist ] Default: index_build |
| --BuildLibraryType | -blt | Library building type, [ Bulk | 10X | Smart-seq2 | inDrop | Drop-seq ] |
| --IndexProjectPath | -ipp | The absolute path of the index-building project (Species name). |
| --Hisat2ThreadNum | -htn | The thread number for hisat2 index-building. Default: 30 |
| --RSEMThreadNum | -rtn | The thread number for RSEM index-building. Default: 30 |
| --ReferenceGenomeFasta | -rgf | The fasta file of reference genome. |
| --ReferenceGenomeGtf | -rgg | The gtf file of reference genome. |
| --StarPath | -sp | The absolute path of the software STAR. |
| --DesignatedAll | -da | The sample list you want to run at once. [ Designated_samples | All_samples ] Default: All_samples |
| --SampleList | -sl | The list samples you designated. [ Sample1-Sample2-Sample3 ] Note: The short dashes are needed. |
| --SequencingType | -st | [ pair | single ] |
| --ReadType | -rt | [ sra | fastq ] Default: fastq |
| --RawData | -rd | The absolute path of raw data |
| --Hisat2Index | -hi | The Hisat2 reference genome index, for example, ../Homo_Sapiens_hisat2/genome |
| --RSEMIndex | -ri | The RSEM reference genome index, for example, ../Homo_Sapiens_RSEM/Homo_Spaiens |
| --BedFile | -bf | The browse extensive data file (.bed file). |
| --FasterqDumpThread | -fdt | The thread number for fasterq-dump. Default: 12 |
| --Fastp_q | -fq | The parameter "-q" in fastp. Default: 20 |
| --Fastp_u | -fu | The parameter "-u" in fastp. Default: 20 |
| --Fastp_l | -fl | The parameter "-l" in fastp. Default: 50 |
| --Fastp_W | -fW | The parameter "-W" in fastp. Default: 4 |
| --Fastp_M | -fM | The parameter "-M" in fastp. Default: 12 |
| --Fastp_w | -fw | The parameter "-w" in fastp. Default: 12 |
| --Hisat2_p | -hp | The thread number for hisat2. Default: 12 |
| --SamtoolsThread | -std | The thread number for samtools. Default: 12 |
| --RSEMThread | -rtd | The thread number for RSEM. Default: 12 |
| --CellrangerIndex | -ci | The absolute path of cellranger index, for example, ../Homo_Sapiens/Homo_Sapiens.genome |
| --CellrangerLocalCores | -clc | Caution! The default localcores may not be appropriate all the time, you can adjust the localcores according to https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/count. Default: 12 |
| --CellrangerLocalMem | -clm | Caution! The default localmem may not be appropriate all the time, you can adjust the localmem according to https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/using/count. Default: 64 |
| --DropTag_p | -dp | The thread number of dropTag process. Default: 12 |
| --StarRunThreadN | -srtn | The thread number of STAR alignment. Default: 12 |
| --DropEst_c | -dc | The configure file (.xml file) of DropEst process. |
| --DropReport_m | -dm | The reference organelle gene's rds file. |
| 长参数 | 短参数 | 描述 |
|---|---|---|
| --workpath | -wp | The absolute path of work path. |
| --exprData | -exp | Expression matrix file, if stream is "all", it would use upstream results. |
| --metaData | -md | Meta information file. |
| --refpath | -rf | Reference file for single cell data. |
| --geneList | -gl | Gene list file. |
| --thread | -td | Number of thread for downstream. Default: 10 |
| --picType | -pic | Picture file format, [ svg | pdf | png ]. Default: svg |
| --rowSums_count_cutoff | -rc | Low count filter. The gene under rowSums count cutoff in samples would be deleted. Default: 2 |
| --pValue_cutoff | -pc | The p value cutoff in diferent analysis. Default: 0.05 |
| --padj_cutoff | -qc | The q value cutoff in diferent analysis. Default: 0.1 |
| --log2FoldChange_Up | -F | Log2(FoldChange) in up regulation cutoff. Default: 1 |
| --log2FoldChange_Down | -f | Log2(FoldChange) in down regulation cutoff. Default: -1 |
| --topnum | -tn | Top number gene cutoff. Using how many top gene as result. Default: 20 |
| --module | -module | Select module to export from colorlevels in WGCNA. Default: turquoise |
| --outputType | -ot | Select export format, [ VisANT | cytoscape ]. Default: cytoscape |
| --nTop | -ntop | Export network cutoff: the number of the top gene to be filtered and exported in selected module. Default: 10000 |
| --NetworkThreshold | -nt | Export Network cutoff: weight > NetworkThreshold. Default: 0.02 |
| --GeneSign | -gs | Hub gene cutoff: significance of single gene-trait correlation > GeneSign. Default:0.1 |
| --absdatKME | -KME | Hub gene cutoff: eigengene connectivity (kME) value > absdatKME Default:0.1 |
| --qWeight | -qw | Weight cutoff. Default:0.9 |
| --GOAnalysis | -GO | GO enrichment analysis, [ yes | no ]. Default: yes |
| --KEGGAnalysis | -KEGG | KEGG enrichment analysis, [ yes | no ]. Default: yes |
| --DOAnalysis | -DO | DO enrichment analysis, [ yes | no ]. Default: yes |
| --ontology | -ontology | GO Ontology of GO enrichment analysis, [ ALL | BP | MF | CC ]. Default: ALL |
| --pValue_cutoff_enrich | -pen | Enrichment analysis cutoff to p value. Default:0.05 |
| --padj_cutoff_enrich | -qen | Enrichment analysis cutoff to q value. Default:0.1 |
| --Biological_Condition | -bc | Choose one case or control, consistent with meta table. Default:Case1 |
| --TreeMethod | -tm | TreeMethod, default, [RF (Random Forests) | ET (Extra-Trees)]. Default: RF |
| --nTrees | -ntrees | Number of trees in an ensemble for each target gene. Default:50 |
| --min_cells | - | The minimum number of cells. Default:5 |
| --max_cells | - | The maximum number of cells. Default:10000 |
| --min_genes | - | The minimum number of genes. Default:200 |
| --max_genes | - | The maximum number of genes. Default:4000 |
| --max_mito | - | The maximum number of mito. Default:0.05 |
| --PCnumber | - | The top PC number for find Neighbor. Default:15 |
| --min_Resolution | - | The minimum Resolution for pre-find cluster. Default:0.6 |
| --max_Resolution | - | The maximum Resolution for pre-find cluster. Default:1.2 |
| --intervals_Resolution | - | The Resolution for pre-find cluster. Default:0.2 |
| --Resolution | - | The Resolution for find cluster. Default:1 |
| --method | - | [ tsne | umap ]. Default:tsne |
| --whichCluster | - | Choose one cluster for find marker and show on TSNE plot, the first cluster label as 0 . Default:1 |
| --ref | - | Choose a reference, default [ 1-6 ], 1 HumanPrimaryCellAtlasData, 2 BlueprintEncodeData, 3 MonacoImmuneData, 4 NovershternHematopoieticData, 5 DatabaseImmuneCellExpressionData, 6 HumanPrimaryCellAtlasData and BlueprintEncodeData. Default:1 |
| --annolevel | - | [ main | fine ]. Default:main |