
- 3. Data analysis standards
- 4. Nomenclature standards
3. Data analysis standards
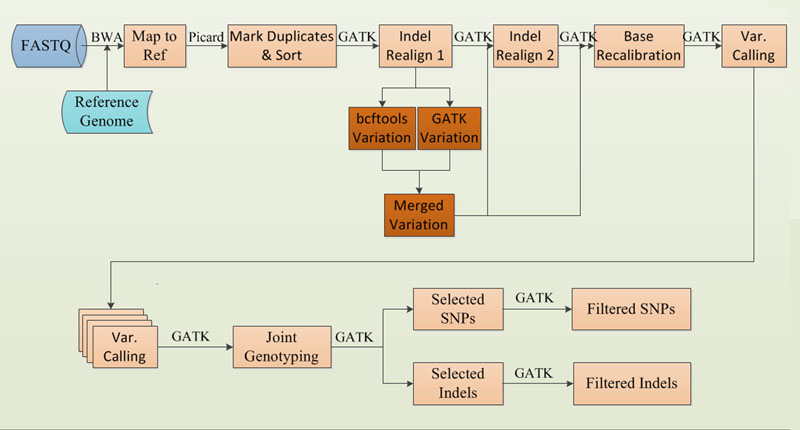
2.For Species without known variations
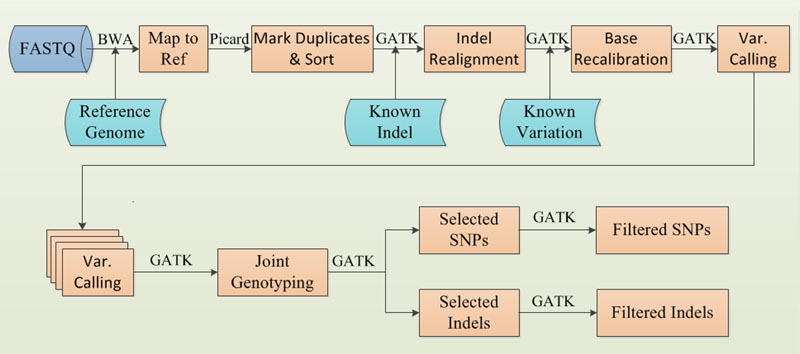
1. Reads mapping to reference genome
bwa mem program is used for genome alignment with the –R and –M parameter. The "-R" flag adds the read group header line and "–M" flag to flag extra alignment hits as secondary.
2. Mark Duplicates & Sort
The software Picrd is employed for marking duplicates and sorting mapping result. MarkDuplicates.jar program of Picard is used for marking duplicate reads and SortSam.jar program is used for sorting the mapping result in coordinate (parameter SORT_ORDER=coordinate).
3. INDEL Realign
The program of "RealignerTargetCreator" and "IndelRealigner" of GATK are used for INDEL Realignment. The "RealignerTargetCreator" identifies what regions need to be realigned, with the known indel sites information. And the "IndelRealigner" performs the actual realignment.
4. Base Recalibration
In the Base Recalibration step, programs "BaseRecalibrator" and "PrintReads" of GATK are employed. Firstly the "BaseRecalibrator" program models the error modes in virtue of the known variation sites, secondly, the "PrintReads" program applies recalibration and writes to file.
5. Variation Calling
Variation calling is performed for individual sample by the "HaplotypeCaller" of GATK. A raw GVCF. format file is created for each sample. In this step, the parameter "-variant_index_type" is assigned LINEAR, the "ERC" is assigned GVCF and the "variant_index_parameter" equals to 12800.
6. Joint Genotyping
Mult-sample GCVF files are jointed genotyping to produce a squared-off matrix of genotypes for analysis. This is implementing by the "GenotypeGVCFs" program of GATK, with multiple single sample GVCF. file as input.
7. Select SNP/INDEL
"SelectVariants" program of the GATK is used for selecting variant for INDEL or SNP with parameter "selecType" sets to INDEL or SNP.
8. SNP Filter
In order to have high quality SNPs, SNP filtering is carried out by the "VariantFiltration" program of the GATK pipeline. The criterion is adopted by the recommending parameters, as "QD<2.0, FS>60.0, SOR>4.0, MQ<40.0, MQRankSum<-12.5, ReadPosRankSum < -8.0".
9. INDEL Filter
In order to have high quality INDELs, INDEL filtering is carried out by the "VariantFiltration" program of the GATK pipeline. The criterion is adopted by the recommending parameters, as "QD<2.0, FS>200.0, SOR>10.0, InbreedingCoeff<-8.0, ReadPosRankSum < -8.0".
10. Prepare reference sites for species without known variation sites
For species without known variations, we need construct solid computational variations to replace. The following are the commands and parameters:
(1) Sample variation calling with GATK
(2) Sample variation calling with samtools and bcftools
(3) Combining of the GATK calling and samtools calling
(4)Hard filter
The criterion is adopted by strict parameters, as "QD<2.0, FS>60.0, SOR>10.0, InbreedingCoeff<-8.0, ReadPosRankSum < -8.0".