The automatic prokaryotic genome annotation pipeline for GWH, based on NCBI's Prokaryotic Genome Annotation Pipeline (PGAP), is designed to streamline the annotation process of prokaryotic genomes. This pipeline integrates various computational tools to analyze genomic sequences, predict gene features, and classify functional annotations automatically. It processes raw genomic sequences by identifying coding regions, non-coding elements, and potential genomic features, ultimately generating comprehensive annotation reports. This automation enhances efficiency, accuracy, and scalability, enabling researchers to focus on downstream analysis and interpretation of genomic data.
We evaluated the quality of each genome reannotation based on the annotated protein results using CheckM. In genome annotation quality evaluation with CheckM, the completeness refers to the proportion of single copy marker genes that are present in the genome to all single copy genes that are specific to a particular taxonomic lineage. Besides the quality of genome sequence, it can also reflect the accuracy of genome annotation using PGAP/userâs pipeline, the higher the better. Contamination, on the other hand, refers to the presence of multiple copies of a single copy gene or foreign sequences in the genome, which can occur due to mis-assembly or contaminated from other organism(s). In the comparison of different annotation results, this value can indicate the detection sensitivity of the genome annotation from another dimension. As of Nov. 1, 2024, there are only 41 genome assemblies with user submitted annotations among all 3925 GWH prokaryotic genome assemblies (Figure 1). The evaluation results of CheckM show that the completeness of 41 PGAP reannotation results is (indicated as mean (+/-standard deviation), the same applies below) around 94.18% (+/-7%), and the contamination rate is around 2.2% (+/-1.87%). The completeness of the 41 user-submitted genome annotation is around 91.72% (+/-0.25%), and the contamination rate is around 1.28% (+/-1.27%). Those data shows that the quality of the PGAP/user submitted gene structure annotations is both high in most cases. But in some cases, there are significant improvement with PGAP reannotation comparing to user-submitted annotation. Moreover, the improvements of the GWH reannotation resource is mainly reflected in the following points: (1) The incorporation of NCBI PGAP to GWH serves as a uniform and standardized pipeline and provides high-quality genome annotations to those assemblies that do not have an annotation (from 0.014% to 92.44%); (2) Besides protein-coding genes, GWH reannotation provides ncRNA, CRISPR elements and fast evolving genes annotations (from 0.008% to 88.98%); (3) In addition, GWH reannotation can also provide identification of pseudogenes (from 0.0002% to 88.95%). (4) Besides gene structure, it provides gene function annotation (from 0.008% to 92.44%). Those improvements will help the users to utilize the genome assembly data and promote genomic research.
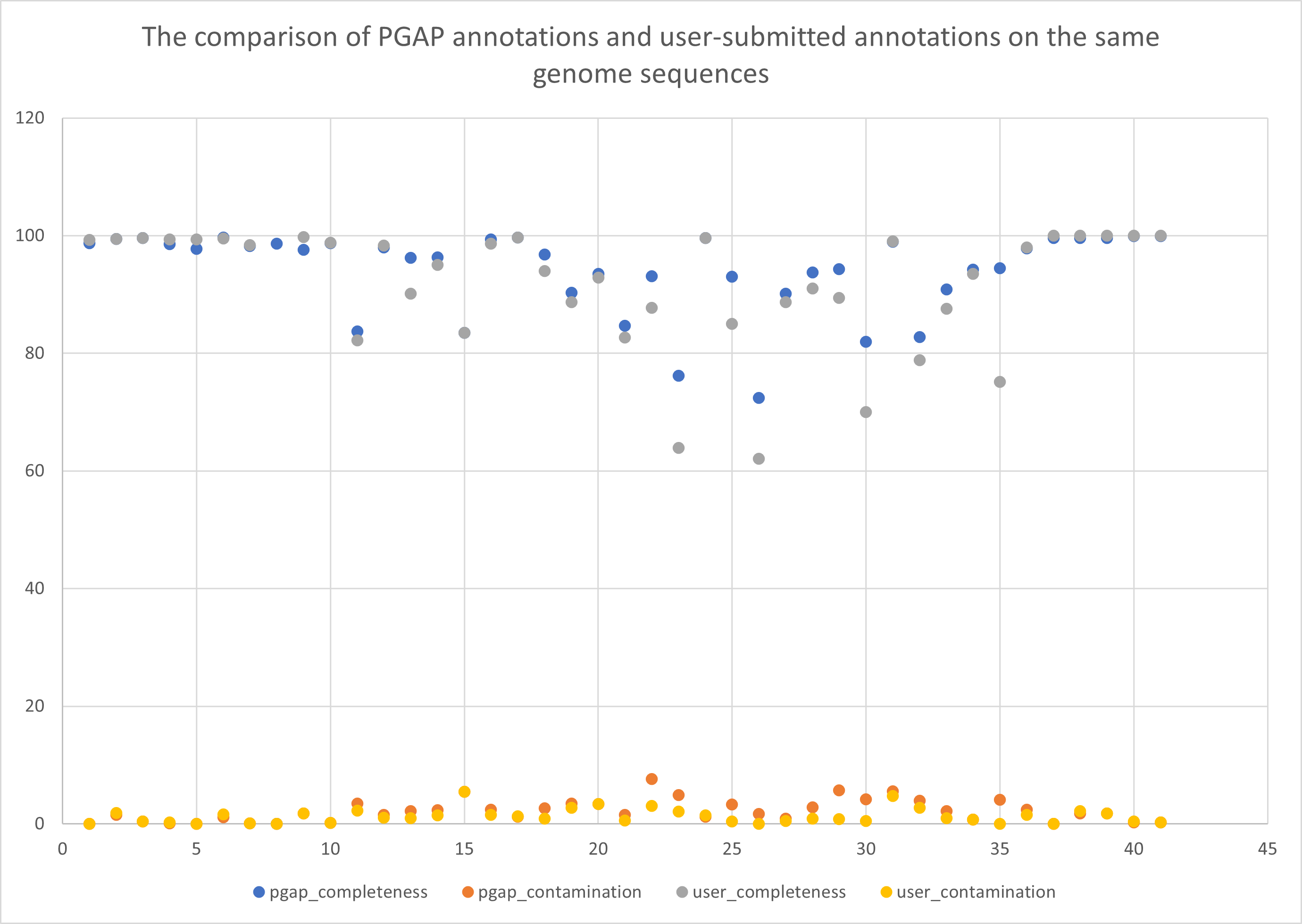
Figure 1 The comparison of PGAP annotations and user-submitted annotations on the same 41 genome sequences.