1. Introduction
1.1 What is LncBook ?
LncRNAs (Long non-coding RNAs) are closely associated with human health and diseases. While the human genome transcribes hundreds of thousands of lncRNAs, only a small part of them have been experimentally studied. The comprehensive annotation of human lncRNAs is of great significance in navigating the functional landscape of the human genome and deepening the understanding of the multi-featured RNA world.
To facilitate the discovery of lncRNAs’ biological functions, we developed LncBook, which is devoted to the integration and multi-omics annotation of human lncRNAs. It provides a comprehensive and high-quality list of human lncRNAs, enriches these lncRNAs with essential multi-omics signatures, and identifies featured lncRNAs in diseases and diverse biological contexts.
The first version was published in the NAR 2019 Database Issue. Over the past several years, we have significantly updated, expanded and enriched LncBook. The updated version of LncBook 2.0 integrates more human lncRNAs, characterizes diverse molecular signatures of these lncRNAs with more abundant data and stringent criteria, and identifies a list of high-confidence lncRNAs that are most likely related with human health and diseases.
1.2 Where does LncBook 2.0 improve?
Compared with the first version, LncBook 2.0 has significant changes and improvements as follows:
2. LncRNA integration and curation
Based on LncBook v1 (4 resources are integrated, including GENCODE v27, NONCODE v5.0, LNCipedia v4.1 and MiTranscriptome beta ), LncBook 2.0 integrates lncRNAs from 5 resources, incuding RefLnc, GENCODE v33, CHESS v2.2, FANTOM-CAT (lv4_strigent) and BIGTranscriptome.
Similar to the first version, we remove transcripts with redundancy, background noise and mapping error, as well as incomplete transcripts, short ones, and those that may encode proteins.
To improve the curation quality, LncBook 2.0 removes lncRNA transcripts without strand information, and transcripts identified as miRNA precursors, small RNAs and pseudogenes according to the comparison results generated with GffCompare.
In addition, four algorithms, viz., CPC2, LGC, CPAT and PLEK, are used for coding potential estimation, and transcripts identified as lncRNAs by at least three algorithms are retained.
However, the high-confidence lncRNAs annotated by HGNC and GENCODE are retained regardless of the coding potential.

The new version of LncBook focuses on lncRNA genes rather than transcripts. Following the strategies used by GENCODE and NONCODE, we assign lncRNA transcripts overlapped in their exonic regions in the same strand into the same gene. Accordingly, all transcripts and annotations are organized by lncRNA genes.

3. LncRNA classification
Based on their genomic locations in respect to protein-coding genes, we classify lncRNAs into seven groups, Intergenic, Intronic (S), Intronic (AS), Overlapping (S), Overlapping (AS), Sense, and Antisense. "S" in the bracket represents that lncRNAs are in the same strand of protein-coding RNAs, and "AS" represents that lncRNAs are in the antisense strand of protein-coding RNAs.

4. Conservation
Based on the UCSC genome alignments between human and 40 vertebrates, we identify homologous protein-coding/non-coding genes and determine gene age for human lncRNA genes.
4.1 Alignment details
We compare the alignments of different transcripts of lncRNA gene, and choose the best-aligned transcript. The lncRNA transcript with the most matched bases in the alignment is considered as the best-aligned transcript, which is used to represent the gene.
Alignment length is the total length of all alignments of the best-aligned transcript.
Alignment identity is calculated as the weighted average by length and identity for all alignments of the best-aligned transcript.
4.2 Sequence conservation
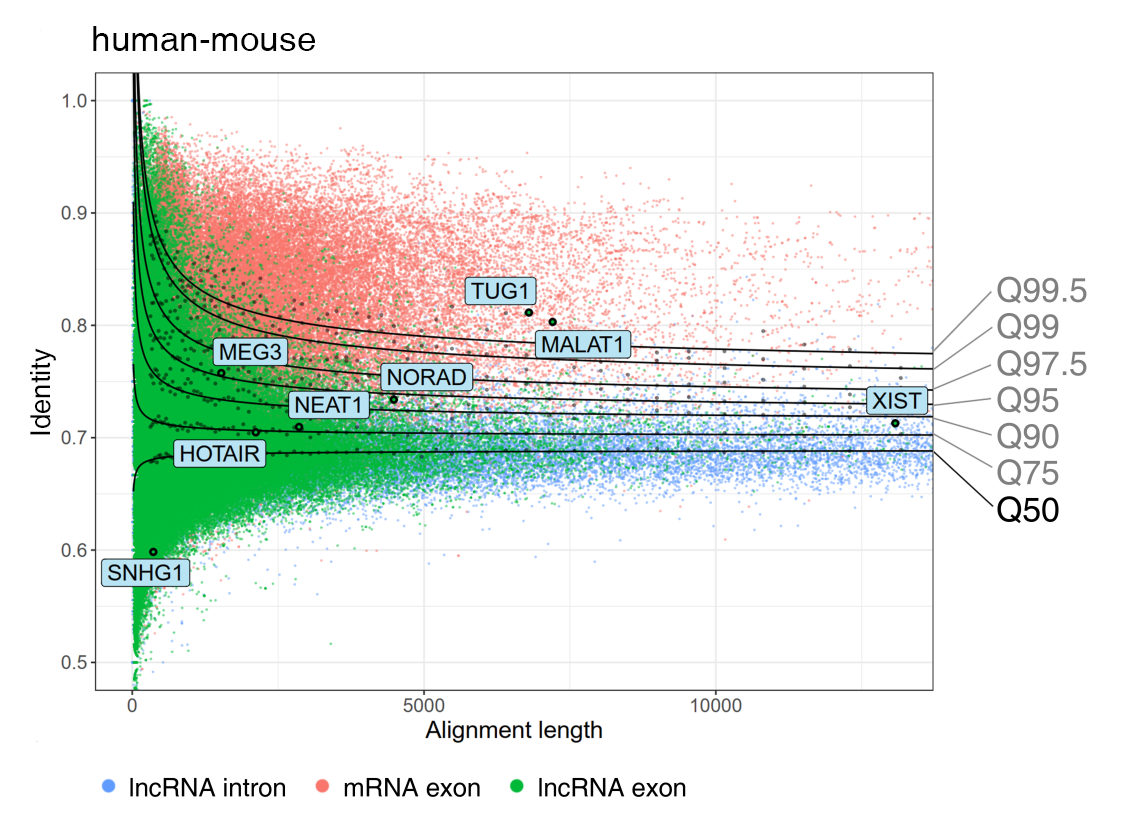
We identify lncRNA homologous sequences/genes by considering alignment length and comparing with introns’ alignments in different species. Specifically, we collect alignments that are at least 50 nt in length and with aligned sequences accounting for more than 20% of the lncRNA transcripts. For a specific species, the homologous sequences/genes are determined if the alignment performance (measured by alignment length and identity) of lncRNAs exceeds the introns’ Q50 threshold, which represents the intermediate level of intron alignments.
For example, in the comparison between human and mouse, mRNA exons are more conserved than lncRNA exons, and lncRNA introns are the least conserved. Among all the lncRNA genes, TUG1 and MALAT1 are highly conserved, whose alignment performances have exceeded the introns' Q99.5 threshold.
Totally, we identify 139,306 homologous genes for 22,347 human lncRNA genes.
4.3 LncRNA age
LncRNA gene age is defined as the earliest occurrence time of homologous sequence, which, from latest to earliest, are "Homo" (human specific), "Hominini", "Homininae", "Hominidae", "Hominoidea", "Catarrhini", "Simiiformes", "Haplorrhini", "Primates", "Euarchontoglires", "Boreoeutheria", "Eutheria", "Theria", "Mammalia", "Amniota", "Tetrapoda" and "Euteleostomi". The 17 time nodes correspond to the 17 important clades of the phylogenetic tree from zebrafish to human.
5. Variation
We collect variants from COSMIC, ClinVar and GWAS Catalog, identify disease/trait-associated variants, annotate corresponding disease/trait information and map them to the lncRNA loci.
5.1 Variants collection and curation
We curate high-quality variants from COSMIC, ClinVar and GWAS Catalog.
5.2 Disease/trait-associated variants
Suggested by COSMIC, the variants whose FATHMM-MKL scores > 0.7 are defined as disease-related (pathogenic) variants. Variants with definite labels such as "benign" or "pathogenic" in Clinvar are collected. For GWAS Catalog, we collected associations with p-value (< 5x10-8), which has been widely used to determine associations between a common genetic variant and a trait of interest.
Names of diseases and traits are unified based on Human Phenotype Ontology and Experimental Factor Ontology, respectively.
5.3 Disease/trait-associated variants allocation
All the disease/trait-associated variants are allocated to lncRNAs by BEDtools intersect function. As a result, we curate 959,138 disease/trait-associated variants associated with 50,165 lncRNA genes.
6. Methylation profile
6.1 DNA methylation data collection
To characterize the DNA methylation profiles of lncRNAs across human diseases, LncBook 2.0 collects 16 publicly accessible bisulfite-seq datasets from TCGA and GEO, covering 14 cancers and 2 neurodevelopmental disorders.
| Biological Context | Source | Project ID | Disease Name (Short Name) | Sample Number |
|---|---|---|---|---|
| Neurodevelopmental Disorder | GEO | GSE119980 | Rett syndrome (RTT) | 12 (6 cases, 6 controls) |
| Neurodevelopmental Disorder | GEO | GSE109875 | Autism spectrum disorders | 16 (10 cases, 6 controls |
| Cancer | GEO | GSE116229 | Acute Lymphoblastic Leukemia (ALL) | 38 (31 cases, 7 controls) |
| Cancer | GEO | GSE135869 | Acute Myeloid Leukemia (AML) | 15 (9 cases, 6 controls) |
| Cancer | GEO | GSE113336 | Chronic Lymphocytic Leukemia (CLL) | 18 (11 cases, 7 controls) |
| Cancer | GEO | GSE149608 | Esophageal Squamous Cell Carcinoma (ESCC) | 19 (10 cases, 9 controls) |
| Cancer | GEO | GSE142241 | Medulloblastoma (MB) | 12 (8 cases, 4 controls) |
| Cancer | GEO | GSE79799 | Liver cancer | 6 (3 cases, 3 controls) |
| Cancer | TCGA | TCGA-BLCA | Bladder Urothelial Carcinoma (BLCA) | 7 (6 cases, 1 control) |
| Cancer | TCGA | TCGA-BRCA | Breast Invasive Carcinoma (BRCA) | 6 (5 cases, 1 control) |
| Cancer | TCGA | TCGA-COAD | Colon Adenocarcinoma (COAD) | 3 (2 cases, 1 control) |
| Cancer | TCGA | TCGA-LUAD | Lung Adenocarcinoma (LUAD) | 6 (5 cases, 1 control) |
| Cancer | TCGA | TCGA-LUSC | Lung Squamous Cell Carcinoma (LUSC) | 5 (4 cases, 1 control) |
| Cancer | TCGA | TCGA-READ | Rectum Adenocarcinoma (READ) | 3 (2 cases, 1 control) |
| Cancer | TCGA | TCGA-STAD | Stomach Adenocarcinoma (STAD) | 5 (4 cases, 1 control) |
| Cancer | TCGA | TCGA-UCEC | Uterine Corpus Endometrial Carcinoma (UCEC) | 6 (5 cases, 1 control) |
6.2 Identification of differentially methylated lncRNAs in 16 diseases
Here, we define the regions from −1500 bps relative to the transcription start site as the promoter region and calculate the averaged methylation level of all CpGs in promoter or body region.
Differentially methylated lncRNAs (featured lncRNA genes) are identified by considering the significance of fold change, p-value, the maximum and minimum values.
Considering the small sampling size of some datasets, different criteria are used.
According to these strict criteria, we identify a total of 19,543 featured lncRNA genes for DNA methylation (hyper/hypomethylated in promoter or body region).
7. Expression profile
LncRNA genes’ expression capacities, featured lncRNA genes across 9 biological contexts (normal tissue/cell line, organ development, preimplantation embryo, cell differentiation, subcellular localization, exosome, cancer cell line, virus infection and circadian rhythm), and expression levels across 337 biological conditions (time point/stage/tissue/cell/component/processing) of the 9 biological contexts are integrated from LncExpDB.
Among 337 biological conditions of 9 biological contexts, genes whose expresion values are greater than the upper quantile of whole transcriptome (includes both lncRNA genes and protein-coding genes) in at least one condition are defined as high-capacity (HC). Low capacity (LC) lncRNA genes are those whose expression values are less than the lower quantiles among all conditions. The remaining lncRNA genes are defined as medium capacity (MC) genes. Genes whose maximum expression values are less than 1.0 TPM in all conditions of a certain context are defined not expressed (NE) genes.
LncRNA genes which are specifically/consistently/differentially/dynamically/periodically expressed in at least on biological context are defined as featured genes.
Accordingly, for each context, the expression capacities are displayed with different colors, and featured genes are represented with the shape of five pointed star in the table of "Expression" section.Collectively, we obtain 29,157 HC lncRNA genes, 48,531 MC lncRNA genes, 8,893 LC lncRNA genes, and 24,070 featured lncRNA genes.
To identify the cell/tissue-specific lncRNAs, we collect the datasets from HPA (32 normal tissues), ENCODE (55 primary cell lines) and CCLE (53 cancer cell lines), and based on the tissue-specific index τ (τ >= 0.9), we identify 40,361 normal tissue/normal cell/cancer cell-specific lncRNAs.
8. Small protein
Small proteins supported by Ribo-seq and mass spectrometry evidence are integrated from SmProt. We map small proteins onto the lncRNAs with BEDtools and those entirely and uniquely fall within lncRNA transcripts are retained. Totally, we integrate 34,012 lncRNA-encoded small proteins for 5,743 lncRNA genes.
9. Interaction
Three tools, including miRanda, TargetScan, and RNAhybrid, are used to predict more lncRNA-miRNA interactions. Interactions supported by all the three tools as well as those by any two tools are retained. Taken together, we predict 146,092,274 lncRNA-miRNA interactions for all lncRNA genes.
Based on the collection of 848,077 RBP (RNA Binding Protein) binding sites of 150 RBPs in HepG2 and K562 cell lines from ENCODE, we map the RBP binding sites onto the lncRNAs with BEDtools and those entirely and uniquely falling within lncRNA transcripts are retained. Totally, we obtain 772,745 lncRNA-Protein interactions for 2,022 lncRNAs.
10. Database usage
10.1 Browse genes
The "Genes" section provides the list of high-quality functional evidences from conservation, gene expression, DNA methylation, genome variation, lncRNA-miRNA interaction and small protein.
Users can obtain the highly conserved lncRNA genes (gene age >= 14) with more multi-omics associations (high expressio capacity, featured gene for expression or methylation, possessing disease/trait-associated variants or encoding small proteins) or query the age and multi-omics associations of a certain lncRNA gene.

10.2 Browse conservation
The Conservation section provides conservation features of human lncRNA genes across 40 vertebrates, as well as alignment details in each species.
You can trace the original gene/sequence of a certain human lncRNA gene, obtain the homologous protein-coding/ncRNA genes in different species, and explore the most conserved or human-specific lncRNA genes.

10.3 Browse variations
The "Variation" section provides all the non-coding variants' genomic location, associated lncRNA, functional effect/clinical significance, and associated diseases/traits information.
You can search interest entries by gene ID, dbSNP ID, functional effect, disease name or trait and download these entries by clicking on download button.

10.4 Browse DNA methylation profile
The "Methylation" section provides all the hyper/hypomethylated lncRNA genes in promoter or body region in at least one disease.
You can explore the methylation status of your interest lncRNA gene across the 16 diseases, or obtain all differentially methylated lncRNA genes in a specific disease.

10.5 Browse expression capacities
The "Expression" section provides all the lncRNA’s expression capacities in various biological contexts.
You can explore high-capacity lncRNAs in one or multiple contexts using the categories in the “Expression Filter”. All entries can be downloaded by click on download button.
Furthermore, the “Chart” enables visualization of expression level distribution among all the biological conditions. Clicking on gene id will direct you to the LncExpDB page where you could view the expression profiles across different biological contexts.

10.6 Browse small proteins
In the "Small Protein" page, you can browse basic information of small proteins, including small protein ID, genome loci, amino acid sequence and experiment evidence.
You can search interest entries by gene ID, small protein ID or experiment evidence and download these entries by clicking on download button.

10.7 Browse interactions
The "Interaction" section provides both lncRNA-miRNA interactions and lncRNA-Protein interactions.
The "LncRNA-miRNA interactions" provides the binding start, binding end, sequence complementarity score and the minimum free energy for the RNA duplexes. You can search interest interactions by gene ID, symbol or miRNA ID and download them by clicking on download button.
The "LncRNA-Protein interactions" provides basic information of the binding start, RBP and cell line. You can search interest interactions by gene ID, symbol, RBP or cell line and download them by clicking on download button.

10.8 Cite LncBook
LncBook 2.0: integrating human long non-coding RNAs with multi-omics annotations (In preparation)
LncBook: a curated knowledgebase of human long non-coding RNAs. Nucleic Acids Res 2019. [PMID=30329098]
10.9 Licenses
LncBook 2.0 is free for academic use only. For any commercial use, please contact us for commercial licensing terms.