What is VISTA
VISTA (VIrus Sequence-based Taxonomy Assignment) is a fast and easy to use tool for analysis of pairwise distance distribution and taxonomic classification for viral genomes within virus families. The distances are pre-computed and stored in the database for every pair within the families and with distribution plotted in a form of histogram where each bar corresponds to an interval of distances.
How to use VISTA
First, go to the list of families and select the family you want to compare with. You can search your interested virus family by Baltimore classification (the type of genome) or directly by their names.
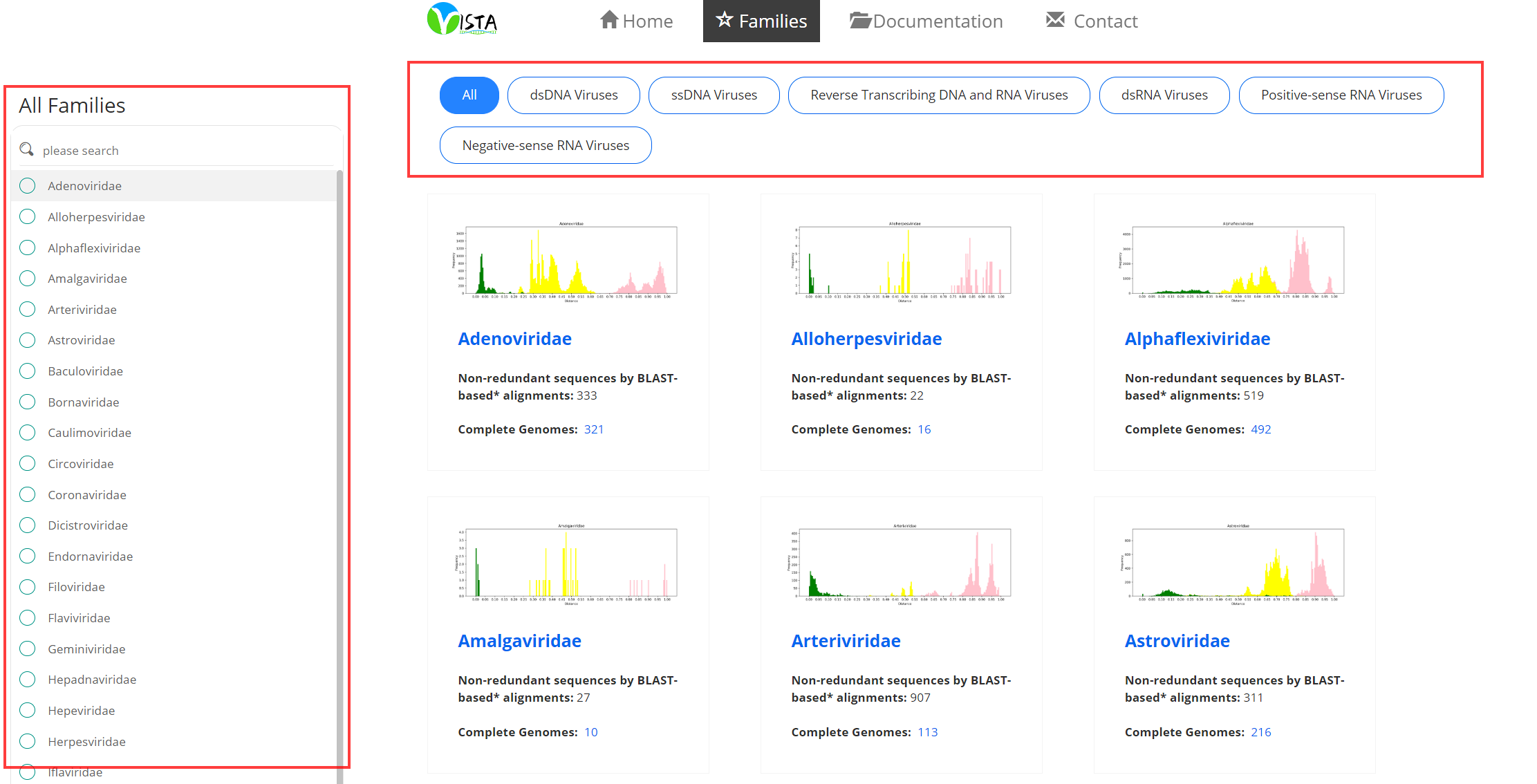
Then, you can see the pairwise distances distribution together with the optimal taxonomic demarcation thresholds on the right side of the page. List of all pairs and distances are in the bottom right position of the page. You can download all data by clicking the green button. Moreover, you can also filter them by taxonomic relationships and distance range.
Genera selection in the bottom left position of the page. The checkboxes allow you to select one or more genera in this virus family and observe the distribution of pairwise distances within or between genera.
If you want to classify newly sequenced genome, the "Upload" box is provided on the left side of the page. You can specify the query genome by pasting it in FASTA format, or by uploading a file containing the sequence using the "Browse" button. After you submit your sequence, VISTA will start calculating pairwise distances between your provided genomes and the existing genome sequences of the family. If the calculations take a long time and you can provide an email address, the assignment result will be sent to you when the calculations are done.
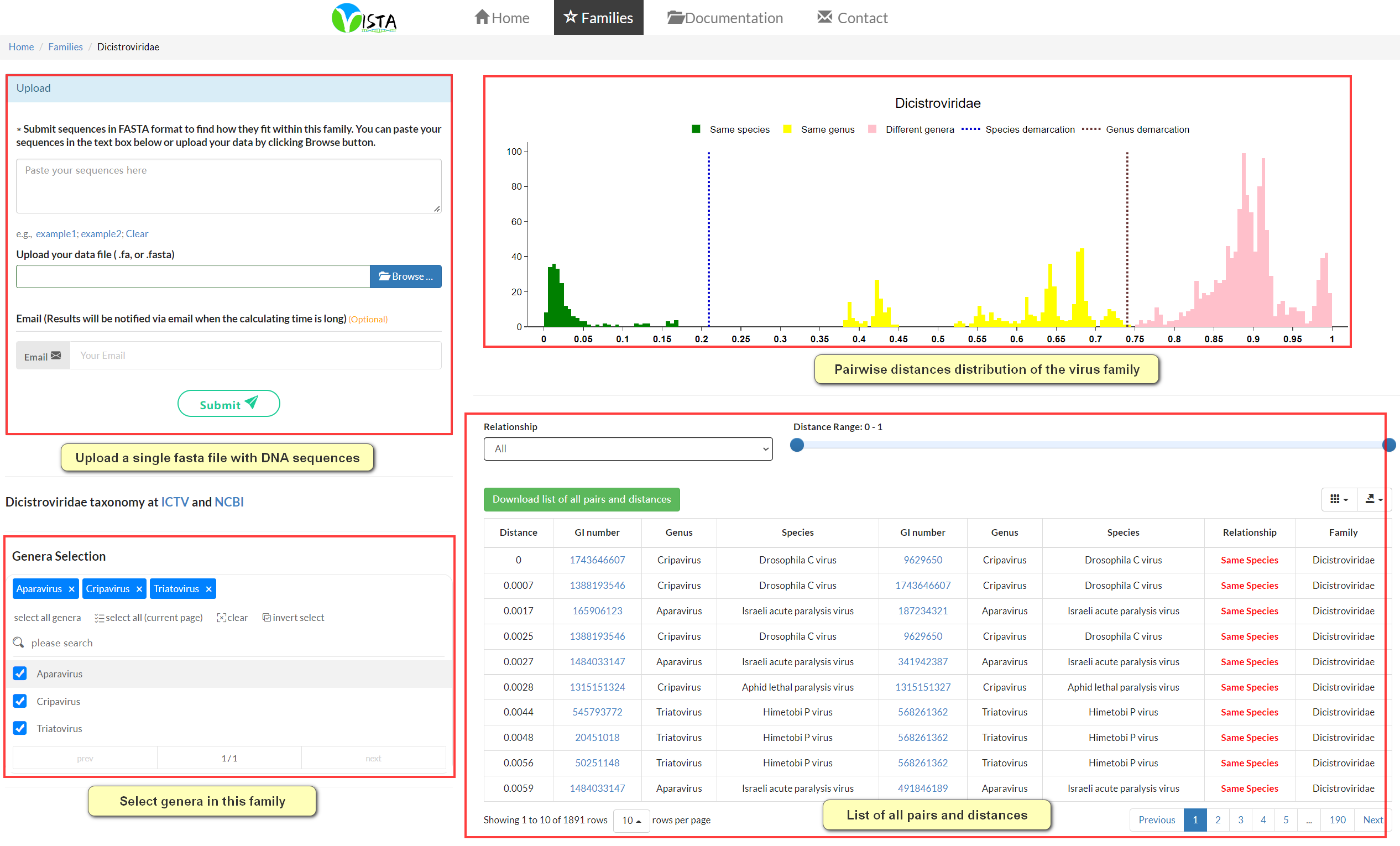
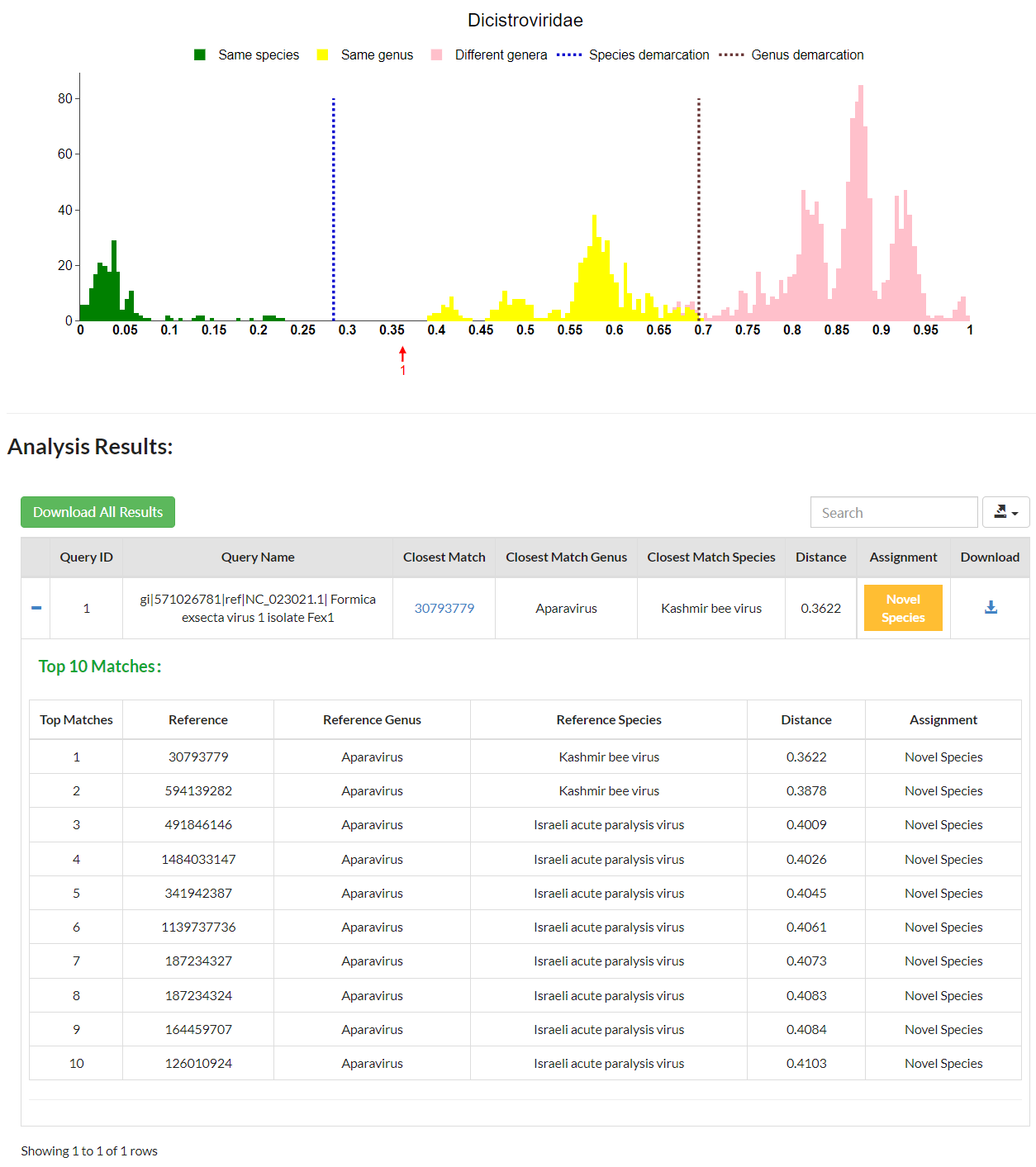
VISTA assignment result
For each input genome, you will be presented with a list of pairwise distances, from the lowest to the highest, between this input genome and 1). the rest of input genomes (if there are more than one), and 2). 10 closest matches to existing genomes within the family. The closest match will be labelled with a red arrow on the distribution graph. VISTA will compare the minimum distance with demarcation thresholds of the relevant family to determine the taxonomic assignment of the input virus. If the minimum distance is below the species demarcation threshold, the input virus could be regarded as a member of the established species showing the lowest distance; if the minimum distance is above the species demarcation threshold but below the genus demarcation threshold, the input virus could be considered as a member of a new species to be created; otherwise, the input virus should be classified as a member of a novel genus. Additionally, the greater the distance from such taxonomic demarcation thresholds, the higher the confidence in the assignment of the input sequence. Users can download all calculated pairwise distances by clicking the green button.
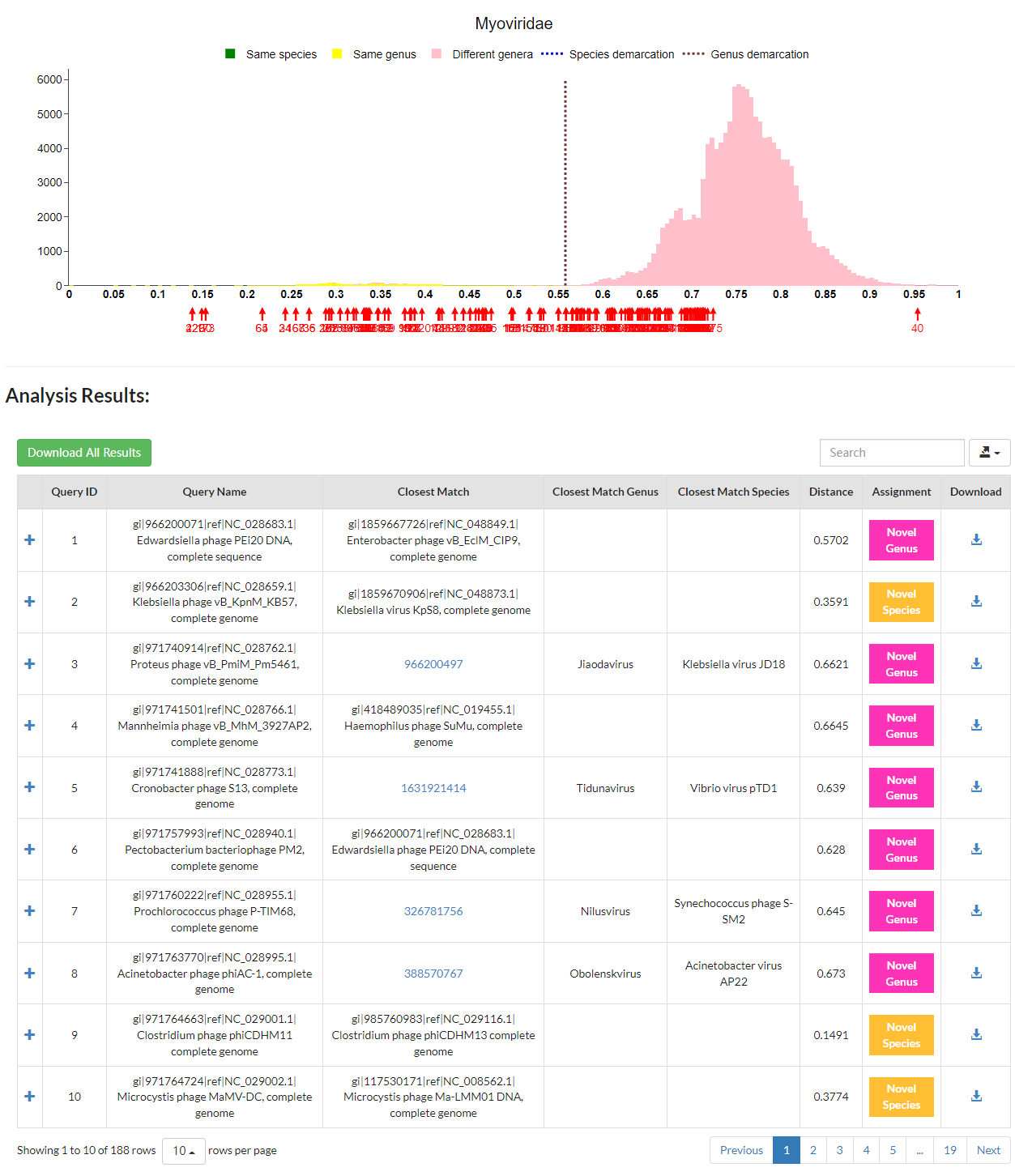
VISTA is able to accept large query sequences.
How to run VISTA locally?
VISTA is also wrapped within a Docker container, with software versions pinned for reproducible execution (https://hub.docker.com/r/taozhangbig/vista). Users can download the Docker images and run VISTA using command lines on the local machine. In Linux OS and Mac OS, once Docker is installed, open a shell window.
1. Pull the docker image using the command:
docker pull taozhangbig/vista:1.0.0
2. Initialize a VISTA container:
docker run -itd --name vista [image_id] /sbin/init
3. Enter the VISTA container:
docker exec -it [container id] bash
4. After VISTA has been installed, you can use "VISTA.sh" in the Scripts directory to start the program:
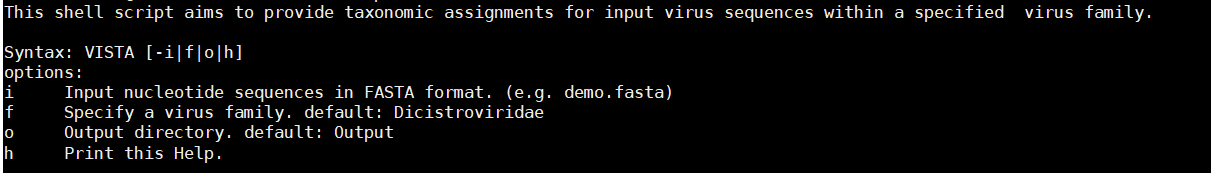
Example:
cd /root/VISTA/
bash Scripts/VISTA.sh -i demo.fasta -f Dicistroviridae -o Output
You can obtain pairwise distances and analysis results in the Output directory.