
 Home
Home
Genome Sequences
Genome Variations
Online Tools
Language / 语言

'Genome tracing' can be used to screen for the closest relatives of the query sequences in the SARS-CoV-2 database and display their spatiotemporal distributions.

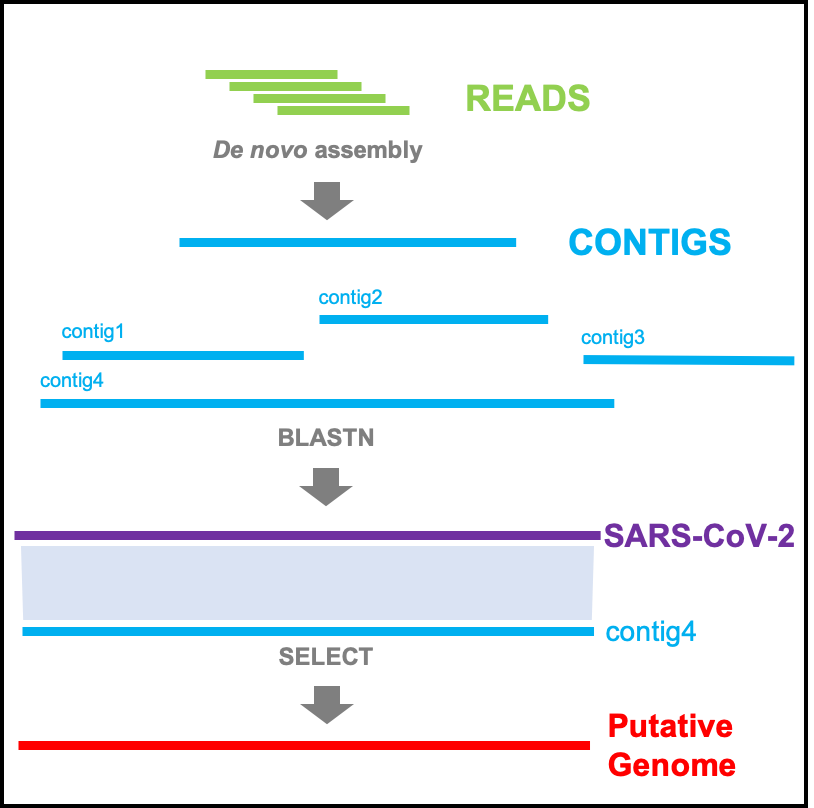
'Denovo Assembly' can be used to assemble NGS sequencing reads,and identify SARS-CoV-2 genome sequences from the assembly contigs.
Using 'BLAST' tool, users can perform sequence alignments with coronavirus genome database, SARS-CoV-2 reference and genome database.

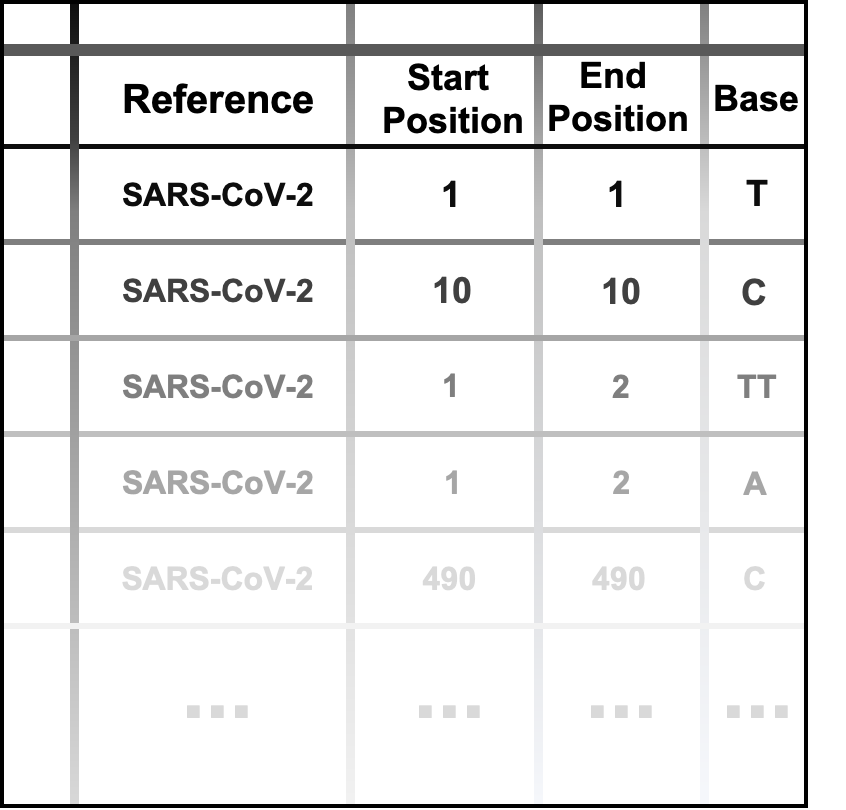
'Genome-to-Variants' can align submitted genome sequences to the SARS-CoV-2 genome and identify SNPs and indels.

'Variant Annotation' can perform functional annotation for the variants, and show the information of genes, code and amino acid changes.
‘Genome Annotation’ supports accurate gene annotations for submitted SARS-CoV-2 genome sequences.
'Fastq-to-Variants' can align NGS sequencing data to the SARS-CoV-2 genome, then identify and annotate SNPs and indels.
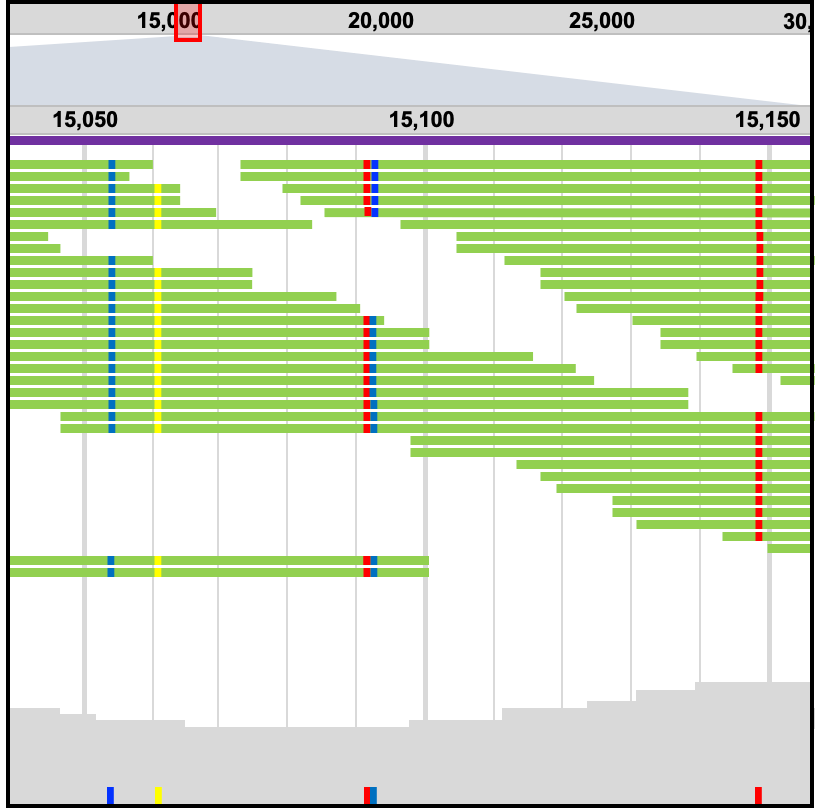
SeqQC can evaluate the sequencing quality of the uploaded FASTQ or BAM files using FastQC and MultiQC.

The haplotype network will be constructed via Haplotype network construction algorithm based on minimum-cost arborescence.
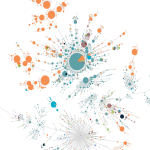
Build Phylogenetic Tree using maximum likelihood method by IQ-TREE or RAxML for the submitted sequences.
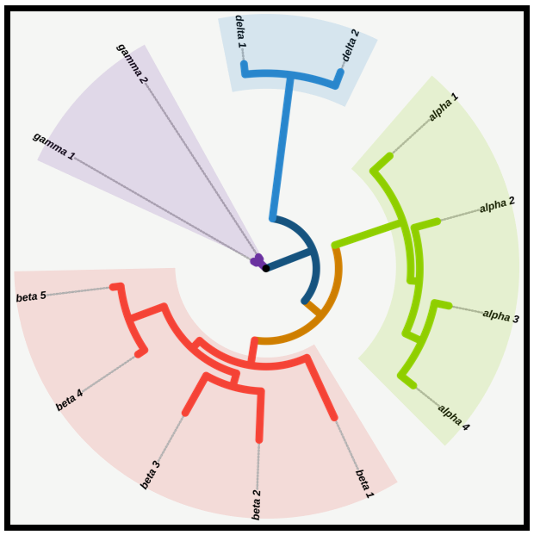
The Pango Lineage Assigner supports Pango lineage assigned for submitted SARS-CoV-2 genome sequences based on the software Pangolin.
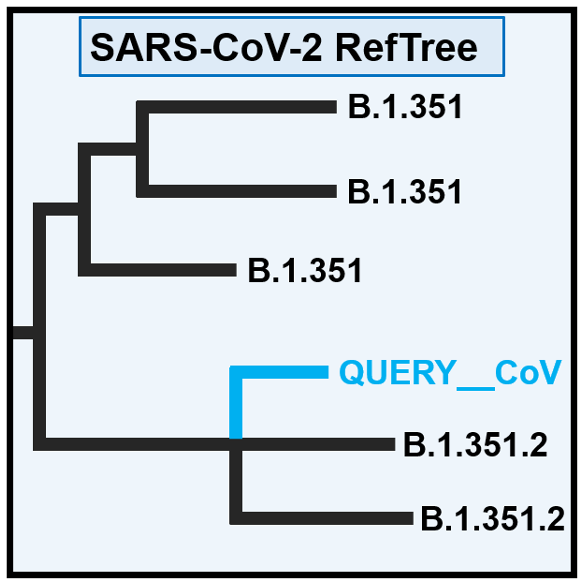
The Pango Lineage Assigner supports Pango lineage assigned for submitted SARS-CoV-2 genome sequences based on the software Pangolin.
