How to use Dog Epigenome Data ?
The Epigenome module integrates data from DNA methylation and ATAC-seq, enabling the exploration of potential regulatory regions within the genome.
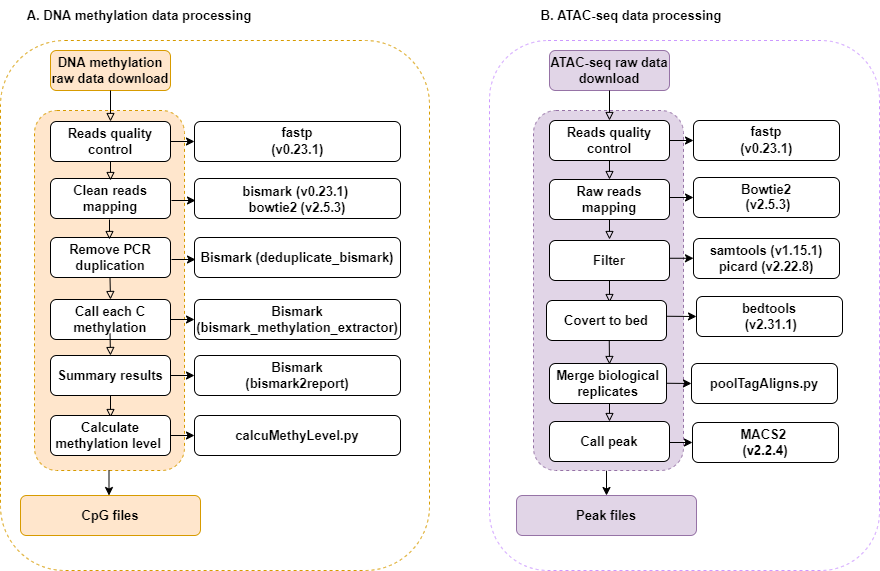
For DNA methylation and ATAC-seq, iDog constructs separate pipelines to process the data. The data analysis code for DNA methylation and ATAC-seq is available for free on GitHub (https://github.com/Br1anChou/idog).
For the analysis of DNA methylation (Supplementary Figure S4A), raw reads were filtered using fastp (v0.23.2) (7) with the parameters ‘g -q 5 -u 50 -n 5’ and then mapped to CanFam4 using Bismark (v0.23.1) (12) with the parameters ‘-g -q 5 -u 50 -n 5’ and then mapped to CanFam4 using Bismark (v0.23.1) (12) with the parameters ‘--bowtie2 --nucleotide_coverage’. Next, PCR duplicates were removed using deduplicate_bismark in Bismark. After calling methylation for each cytosine with bismark_methylation_extractor using the parameters ‘-s --no_overlap --bedGraph --cytosine_report --CX_context’, a methylation call report was produced using bismark2report in Bismark. Finally, the methylation levels (methylated_count/region_length) were quantified using calcuMethyLevel.py (https://github.com/Br1anChou/idog).
For the analysis of ATAC-seq (Supplementary Figure S4B), raw reads were filtered using fastp (v 0.23.2) (7) with the parameters ‘-g -q 5 -u 50 -n 5’. The filtered reads were aligned to the reference using Bowtie2 (v2.5.3) (13) with default parameters. Unmapped reads, low mapping quality reads (≤30), secondary alignments, and PCR duplicates were filtered out using SAMtools (v1.15.1) (14) and Picard (v2.22.8, https://broadinstitute.github.io/picard/). After merging biological replicates from the same samples with poolTagAligns.py (https://github.com/Br1anChou/idog), peaks of accessible chromatin were identified using the callpeak function in MACS2 (v2.2.4) (15) with the following parameters: ‘-p 0.01 --shift -100 --extsize 200 --nomodel -B --SPMR --keep-dup all --call-summits’.
For the analysis of DNA methylation (Supplementary Figure S4A), raw reads were filtered using fastp (v0.23.2) (7) with the parameters ‘g -q 5 -u 50 -n 5’ and then mapped to CanFam4 using Bismark (v0.23.1) (12) with the parameters ‘-g -q 5 -u 50 -n 5’ and then mapped to CanFam4 using Bismark (v0.23.1) (12) with the parameters ‘--bowtie2 --nucleotide_coverage’. Next, PCR duplicates were removed using deduplicate_bismark in Bismark. After calling methylation for each cytosine with bismark_methylation_extractor using the parameters ‘-s --no_overlap --bedGraph --cytosine_report --CX_context’, a methylation call report was produced using bismark2report in Bismark. Finally, the methylation levels (methylated_count/region_length) were quantified using calcuMethyLevel.py (https://github.com/Br1anChou/idog).
For the analysis of ATAC-seq (Supplementary Figure S4B), raw reads were filtered using fastp (v 0.23.2) (7) with the parameters ‘-g -q 5 -u 50 -n 5’. The filtered reads were aligned to the reference using Bowtie2 (v2.5.3) (13) with default parameters. Unmapped reads, low mapping quality reads (≤30), secondary alignments, and PCR duplicates were filtered out using SAMtools (v1.15.1) (14) and Picard (v2.22.8, https://broadinstitute.github.io/picard/). After merging biological replicates from the same samples with poolTagAligns.py (https://github.com/Br1anChou/idog), peaks of accessible chromatin were identified using the callpeak function in MACS2 (v2.2.4) (15) with the following parameters: ‘-p 0.01 --shift -100 --extsize 200 --nomodel -B --SPMR --keep-dup all --call-summits’.
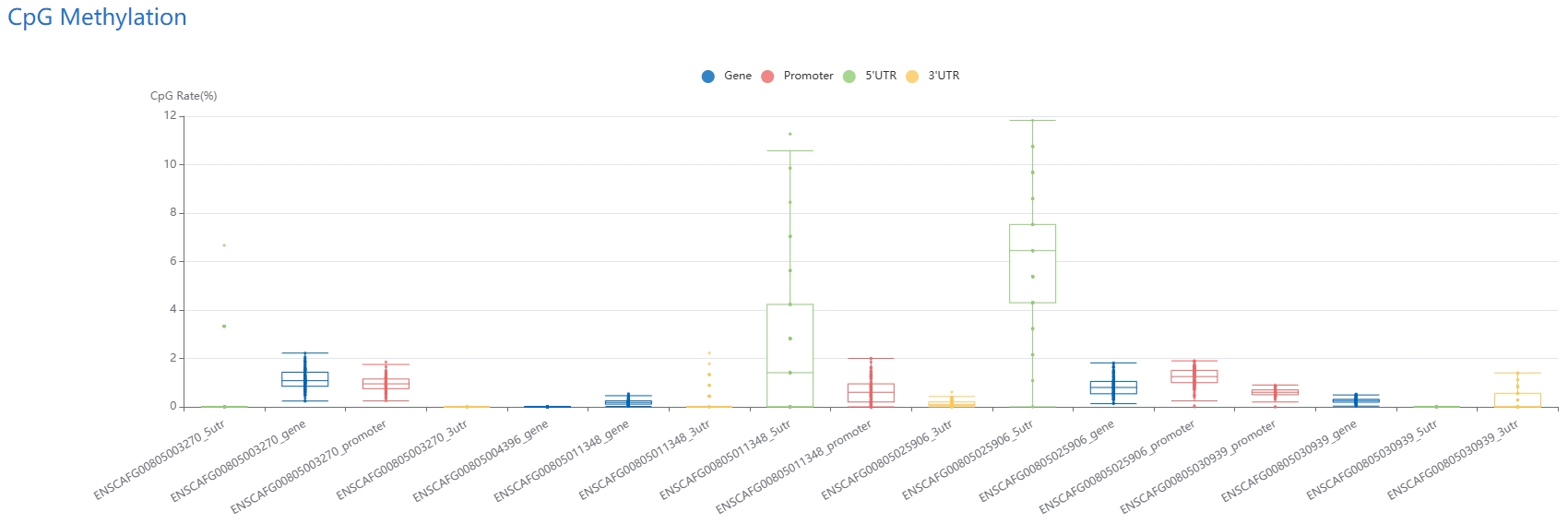
1. DNA methylation
For DNA methylation, during result retrieval, both an average CpG score heatmap and a detailed table with comprehensive CpG information are provided for selected tissues
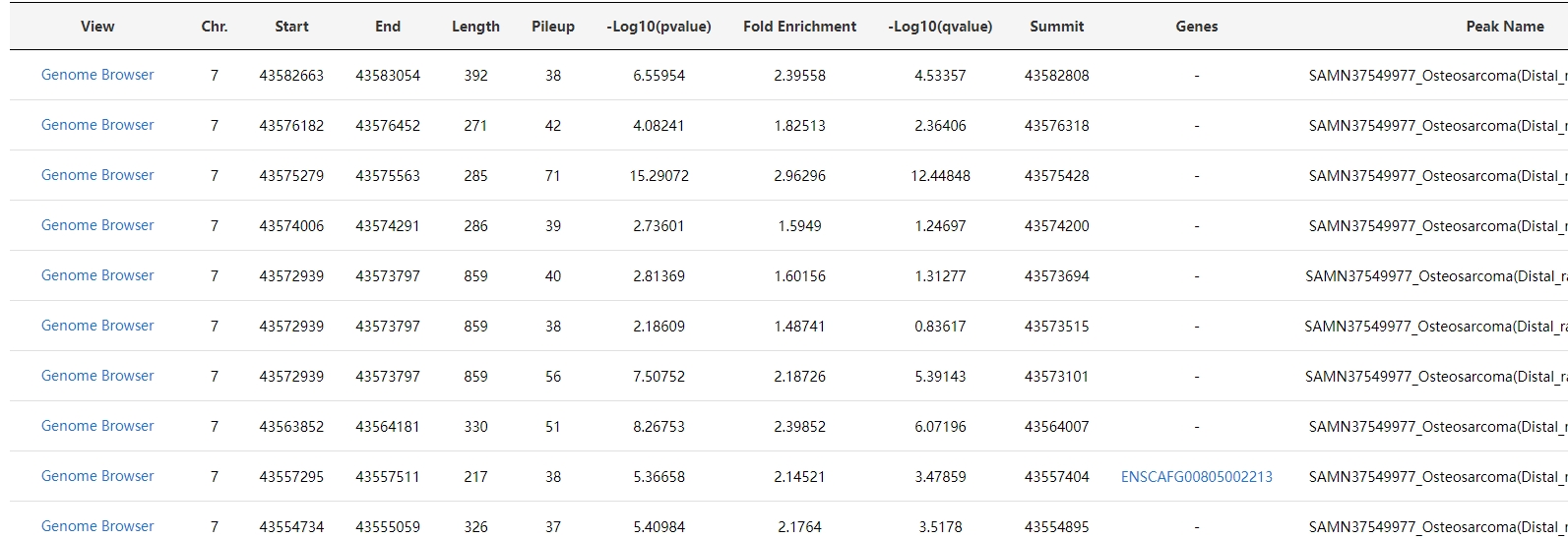
2. Chromatin accessibility
For chromatin accessibility, a genome browser displaying peaks and a table with detailed peak information are presented.
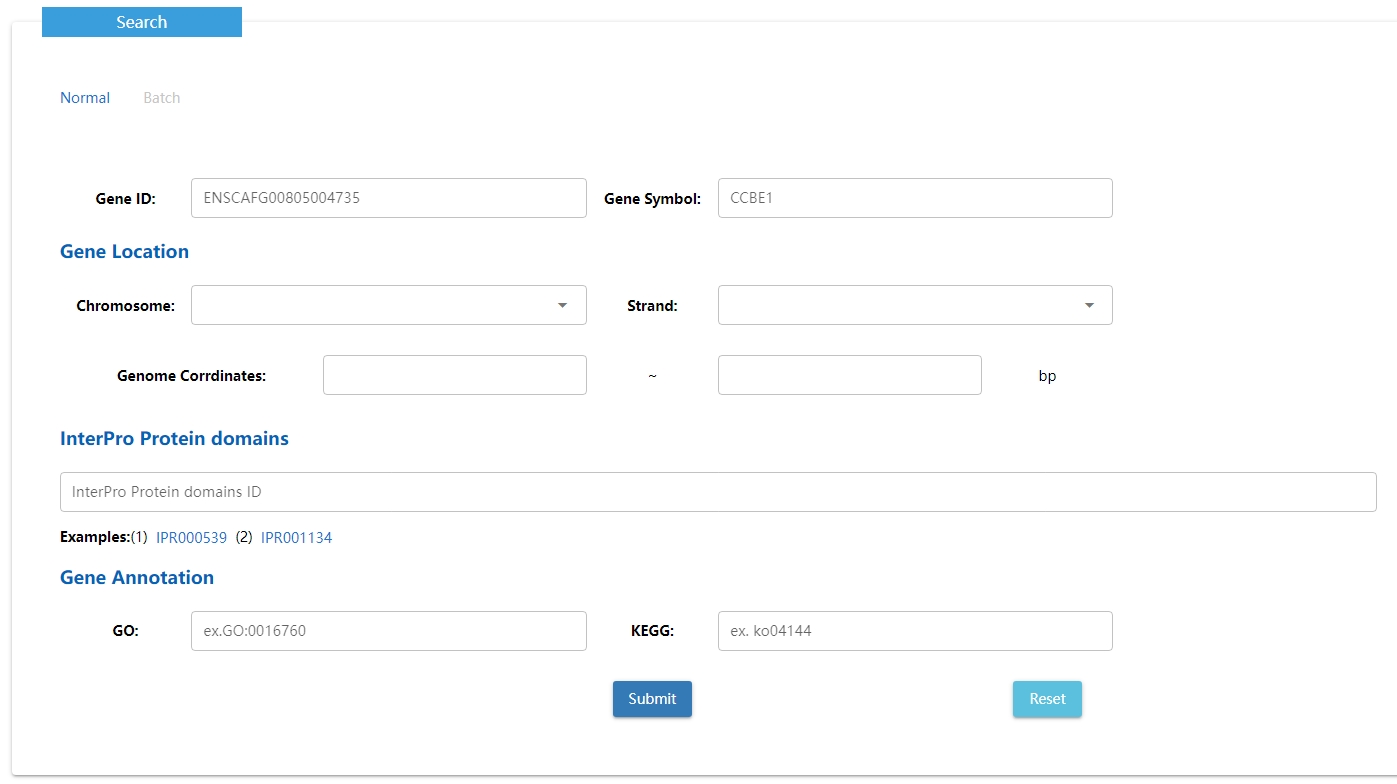
3. Search data
Users can search DNA methylation by various situations including gene, gene location ,InterPro Protein domains and GO annotation term. A batch search for input a gene list is also available.