
Since the rise of single-cell RNA sequencing technology in 2009, the era has witnessed it's rapid development. Benefit from the unprecedented single cell resolution, researchers now have a powerful tool to deeply parse the cell heterogeneities and their inner molecular mechanisms during the whole processes of reproduction, individual development, immunoregulation and carcinogenesis, etc. Therefore, a large number of single-cell RNA sequencing projects were consecutively carried out, and exponential data have been accumulated over the past decade. Meanwhile, several single cell databases were also developed for the integrated mining of these valuable data.
In view of the different origin, different tissue source, different analytical pipeline of these huge volumes of single-cell RNA-seq data, it was challenging for researchers to effectively integrate them and thereby extract valuable knowledge. Facing the high incidence of diverse cancers worldwide, the starting point of this database was to widely collect currently available single-cell RNA-seq datasets for different human cancer types, and further studied the immune profiles and gene expression dynamics in the specific tumor microenvironments. Thus, we can compare the cell components and the expression of many functional molecules between different cancer types, and ultimately dig out some novel superior immune checkpoints which can be used in the future clinical immunotherapy for the particular types of cancer.
Metabolism is the sum of a series of life-giving chemical processes within a cell. It is the final result of gene transcription affected by various internal and external factors, and can directly reflect the physiological phenomena in organisms. Cancer is a heterogeneous disease characterized by various genetic and phenotypic abnormalities. The infinite proliferation of malignant cells creates a huge energy demand, requiring metabolic changes to meet the energy and metabolite needs, which also helps drive tumor development. The process of tumorigenesis is usually accompanied by changes in metabolic patterns, also known as metabolic reprogramming. Research on tumor metabolism reprogramming helps us better understand how these behaviors support tumor growth and determine which reprogramming activities are most relevant to therapeutic outcomes. The transcriptome was widely used to measure tumor metabolism indirectly in recent years. Therefore, a series of standardized single-cell metabolic analyses based on scRNA-seq data were performed and included in CancerSCEM 2.0. By illustrating the distribution of our inferred raw metabolic flux and their dynamic variation at the single-cell resolution, and providing their metabolic correlation patterns as well, users are significantly supported to better understand the particular metabolic landscape of human cancers, and to further perform the carcinogenic mechanism research of some specific cancer types.
Through the widely survey of currently available omics databases, single-cell databases and public literatures, CancerSCEM has collected the raw sequencing data or expression matrix of hundreds of cancer related single-cell RNA-seq datasets from NCBI (GEO), ArrayExpress (EBI), Single Cell Expression Atlas - EBI, Panglaodb, CancerSEA, Single Cell Portal - Broad Institute, SCPortalen and several literatures about cancer single-cell researches etc. Figure 1 on the right side has shown the data summary including raw data sources, the number of high-quality cells in each sample (orange histograms on the outer circle with the maximum equal to 43,913) and the abbreviation of their respective cancer types.
In summary, the single-cell RNA-seq data of a total of 1,104 cancer samples, 304 normal samples and 58 PBMC samples were collected and all the analytical results were cataloged into the database. They have covered a total of 74 human cancer types, and 8 construction protocols have been used. After the sequencing read and cell quality control by filtering low quality cells with significantly abnormal gene expression levels or high mitochondria RNA percentages, a total of 7,320,070 high quality cells have been reserved.

Figure 1. Statistics of cancer single-cell RNA-seq datasets in CancerSCEM 2.0.
CancerSCEM has been working on widely collecting single-cell RNA-seq datasets for various types of human cancers, and performing multi-level analysis on each dataset. Similar to the CancerSCEM 1.0, tailored workflows should be established for the data from different construction protocols so that to obtain the optimal analytical results. As 10X Genomics platform has its own auxiliary processing software Cell Ranger, we thus adopted Cell Ranger v7.1 to handle with all 10X Genomics datasets. The remaining datasets from all other protocols like Smart-seq2 and Drop-seq were all processed by zUMIs v2.9.7d (reads mapping with STAR).
After the gene expression matrix (UMI counts) was generated, the R package DoubletFinder v2.0.2 was applied to doublets removal, and a widely used package Seurat v4.3.0 was utilized to perform cell quality control, PCA dimension reduction, tSNE and UMAP clustering with personalized principal component numbers and clustering resolutions, etc. Next, scCancer v2.2.0, a newly-developed database Cell Taxonomy, SingleR v1.4.1 combined with manual annotations using dozens of marker genes(Table 1) were paralleled used to identify malignant cells, subtypes of immune cells and several other cell types in each dataset. Finally, the 'FindMarkers' function in Seurat was ultilized to perform differential gene expression analysis for each specific cell type, and GO and KEGG enrichments were further performed. CancerSCEM additionally collected hundreds of key functional molecules including receptor genes, ligand genes, oncogenes and tumor suppressor genes from multiple data sources like CelltalkDB, SingleCellSingalR, Cellinker, Cell-Cell Interaction Database, Cancer Gene Census, OncoKB, Network of Cancer Genes, TSGene, IntOGene, etc, and their expression patterns would be shown on the general analysis page for sample.
In particular, a single-cell metabolic map was newly established based on scRNA-seq data in CancerSCEM 2.0. An advanced flux estimation tool scFEA v1.1.2 covering 168 metabolism modules was employed to accurately infer single-cell metabolic flux, and we further manually aligned these modules to the KEGG pathways, obtaining 34 reliable pathways as a consequence. In view of the batch effects across different experiments, the dynamic metabolic variation at the cell-type level was evaluated by the Log2FoldChange value (the mean flux of the given cell type compared with the sample mean). In addition, several important metabolic pathways inlcuding Glycolysis/Gluconeogenesis (hsa00010), TCA cycle (hsa00020), Purine metabolism (hsa00230) and Pyrimidine metabolism (hsa00240) attracted close attention by investigating their metabolic level comparison and metabolic correlation. Correspondingly, a newly-created METABOLIC module including 3 analytical functions was equipped in our interactive online analyze platform.
In downstream, cell-cell interaction networks were built by CellphoneDB, survival analysis of the same or similar cancer types have been performed based on the bulk RNA-seq data and clinical survival data from TCGA. In CancerSCEM 2.0, several new relevant analyses such as transcriptional factor enrichment, copy number variation evaluation and pseudotime trajectory construction were performed and provided in the interactive online analyze platform. Figure 2 has shown the overview of the data processing in CancerSCEM 2.0.
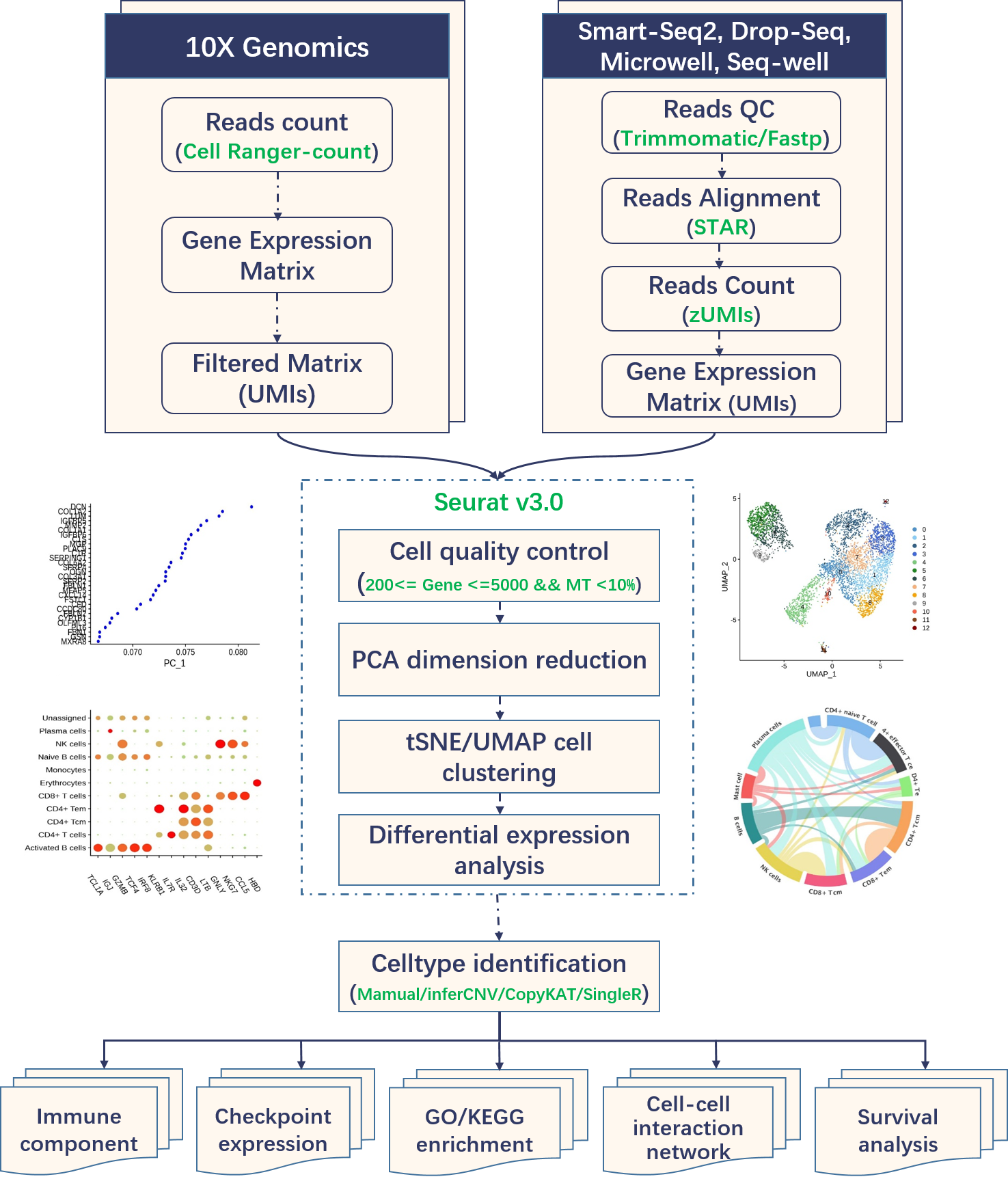
Figure 2. Overview of the data procession for 10X Genomics datasets and other datasets from Smart-seq2, Drop-seq, etc.
Table 1. Cell-type specific marker genes used by CancerSCEM
| Cell type | Cell-type specific markers |
|---|---|
| Astrocyte | AGXT2L1, GFAP, ALDOC, SLC1A3, AGT, ALDH1L1 |
| B cell | CD19, MS4A1, BANK1, BLK, IRF8, ABCB4, ABCB9, AFF4, AIDA, AIM2 |
| Endothelial cell | VWF, PECAM1, CDH5, VEGFA, FLT1, ECSCR, ACYP1, ADGRL2, SELE, ICAM1 |
| Epithelial cell | CDH1, MYLK, ANKRD30A, ABCA13, ABCB10, ADGB, SFTPB, SFTPC |
| Erythrocyte | ALAS2, CA1, HBB, HBE1, HBA1, HBG1, GYPA |
| Fibroblast | COL1A1, COL3A1, THY1, NECTIN1, FAP, PTPN13, C5AR2, LRP1 |
| GMP | CD38, KIT, ADK, CD123, ALDH4A1, ANXA1, AP3S1, APLP2, APPL1, AREG, ASPM, CDKN3, CLSPN, DEPDC7, MCM10, MUCB2, SDC4, RMI2 |
| HSC | CD34, ITGA5, PROM1, CD105, VCAM1, CD164, THY1, KIT, ACE, CMAH, ABCG2, CD41, ALDH1A1, BMI1 |
| Macro/Mono/DC | CD68, CD14, MRC1, BHLHE40, CD93, CREM, CSF1R, CCL18, ICAM4, ACPP, ACSL3, ADGRE2, ADGRE3, CD209, CD83, CD1A |
| Malignant cell | EPCAM, FOLH1, KLK3, KRT8, KRT18, KRT19 |
| Mast cell | SLC18A2, ADIRF, ASIC4, BACE2, ENPP3, CADPS, CAPN3, CDK15, CMA1, GCSAML, MAML1, MAOB, CAVIN2 |
| Melanocyte | MLANA, PMEL, DCT, KIT, MITF, TYR, MC1R, OCA2, BCL2 |
| Muscle cell | MYH2, MYL2, ACTA1, CKM, MYOM3, CRYAB, CMD, APLNR, HOMG4 |
| Myeloid cell | PTPRC, CD14, AIF1, TYROBP, CD163 |
| Neural progenitor cell | SOX2, NESTIN, DCX, NES1, PAX6, CENPF, UBE2C, TMN2, MKI67 |
| Neuron | STMN2, RBFOX3, MAP2, TUBB3, CSF3, DLG4, ENO2 |
| Neutrophil | ADGRG3, CXCL8, FCGR3B, MNDA, USP10, CSF3R, ANXA3, AQP9, BTNL8, LGALS13, G0S2, NFE4, IL5RA |
| NK cell | FCGR3A, KLRB1, KLRD1, NKG7, XCL1, XCL2, NCR3, NCR1, CD247, GZMB, KLRC1, KLRK1 |
| Oligodendrocyte | MOG, OLIG1, OLIG2, PDGFRA, PLP1, MBP, MAG, SOX10 |
| Oligodendrocyte precursor cell | PDGFRALPHA, OLIG2, CSPG4, OLIG1, VCAN, LHFPL3, GPR17, APOD |
| Pericyte | CSPG4, PDGFRB, MCAM, RGS5, ALPHA-SMA, KCNJ8, PDGFRALPHA, THY1 |
| Plasma cell | MZB1, BRSK1, AC026202.3, JSRP1, LINC00582, PARM1, TAS1R3 |
| Plasmacytoid dendritic cell | CLE4C, IL3RA, LILRA4, GZMB, JCHAIN, IRF7, TCF4, NRP1, IRF8 |
| Progenitor | CD38, CASR, ALDH, CAR, KDR, MME, FLT3, CD90, CD123 |
| Retinal ganglion cell | RBPMS, NESTIN, PACA, GAP43, MAP2, ATOH1, NEFM, NEFH |
| Smooth muscle cell | TAGLN, MYH11, DES, CNN1, RGS5, MYL9, NOTCH3, SYNPO2, PLN |
| T cell | CD3D, CD3G, CD3E |
| Treg | IL2RA, FOXP3, CTLA4, LRRC32, TNFRSF18, TNFRSF4 |
Table 2. Metabolic pathways and modules used by CancerSCEM
| KEGG pathway | Module ID | Compound IN name | Compound IN ID | Compound OUT name | Compound OUT ID |
|---|
Tutorial of CancerSCEM can be found here.
Once clicking on the 'Project Browse' button in the navigation bar, an overview table of all collected cancer single-cell RNA-seq projects will be shown, with the information ranging from unique project ID, cancer type, project hold country, sample ID, sample details, cell count to library construction protocol, the last two characters in the newly assigned sample ID respectively represented 1A - 10X Genomics, 1B - Smart-seq2, 1C - Drop-seq, 1D - Microwell and 1E - Seq-Well. Moreover, the 'Sample Details' and 'Analysis' columns in the table will provide hyperlinks to the detailed information of the tumor sample and its general analysis results for each dataset, respectively.
According to the order of the left-hand navigation on the general analysis page, multiple levels of analytical results would be presented including 'Data Statistics and tSNE/UMAP Visualization', 'Tumor Microenvironment' and 'Functional Genes' Expression' (receptor genes, ligand genes, oncogenes and TSGs). All results are shown in tables or figures, and several cell types are abbreviated as follows: CD4+ central memory T cells (CD4+ Tcm), CD4+ effector memory T cells (CD4+ Tem), CD8+ central memory T cells (CD8+ Tcm), CD8+ effector memory T cells (CD8+ Tem), Regulatory T cells (Tregs), Natural killer cells (NK cells), Hematopoietic stem cells (HSCs), Granulocyte macrophage progenitors (GMPs).
CancerSCEM provided several search channels as follows:
a) Quick search on the home page: provided user real-time querying service merely by cancer type, gene symbol or gene ID. The corresponding projects/samples with their analysis results and the overall expression patterns of the target gene would be generated.
b) Advanced search on the search page: there are 4 modules user can utilize to seek for their interested projects/samples or specific genes. For projects, user can specify a definite project ID, sample ID or a known accession No., or select a particular cancer type (abbreviation) or a construction protocol, the overview and also details of the target projects/samples will thus been obtained. For genes, inputting a gene symbol or gene ID would trigger an instant query to the database, in return its expression profiles in both single-cell and bulk RNA-seq level to endpoint user.
c) Keyword cloud was also provided on the home page, each word will link to a browse or analysis page with the detailed information of the selected word, it's highly intuitive and easy-to-use for user.
Online analyze is the most characteristic platform in the database. A total of 4 analyze modules are equipped: GENE, SAMPLE, CELL and METABOLISM, the latter two of which were newly created in CancerSCEM 2.0. GENE module mainly focused on the 1) Gene Expression (GE) in Sample - whole expression profiles of target gene in specified cancer single-cell sample and 2) GE in Subtypes - it's expression in different cell subtypes in the sample, 3) GE Correlation - gene expression correlation analysis in the specific sample and 4) GE Comparison - expression comparison between different single-cell RNA-seq or TCGA bulk RNA-seq datasets. SAMPLE module included three analyze functions: 1) Cell Component Comparison - cell type component comparison between single-cell samples, 2) Cell Interaction - interaction network construction between different cell types and 3) Survival Analysis - survival analysis based on TCGA bulk RNA-seq data and clinical survival data. CELL module has deployed three analytical contents: 1) The visualization of genomic copy number variation across different cell types especially for the Malignant cells, 2) The enrichment of transcription factors for each cell type, 3) An inferred pseudotime trajectory in the target cancer sample and 4) Seven biological features scoring, including cytotoxicity, HLA-dependent or -independent activating or inhibitory receptors, inflammatory response, and stress.
For the purpose of supporting users to perform personalized data mining on metabolism, the visualization of 1) the distribution of raw metabolic flux, 2) dynamic variation (Log2FoldChange) and 3) the metabolic correlation at both the module and pathway hierarchies are available in the newly-created METABOLISM module. Benifit from this module, users are significantly supported to better understand the particular metabolic landscape of human cancers, and to further perform the carcinogenic mechanism research of some specific cancer types.
The interactive analyze platform in CancerSCEM 2.0 containing 4 modules and 13 functions has covered almost the full spectrum of scRNA-seq data analysis (Figure 3). No matter in which analyze moduel, user need to specify the unique sample ID, a gene symbol or gene ID, and several alternative parameters like figure colors and the number of cell interaction pairs are also supplied. All analytical results could be displayed in real time.

Figure 3. The structure overview of the Online Analyze Platform in CancerSCEM 2.0.
Original metadata, raw UMI counts matrix (users are feasible to perform some personalized analysis by this), cell component for each single-cell dataset and differential expression gene list for each cell subtype are available for download, and most of the figures and tables in analysis page can be exported to user's local computer.