Home
Home
Genome Sequences
Genome Variations
Online Tools
Language / 语言

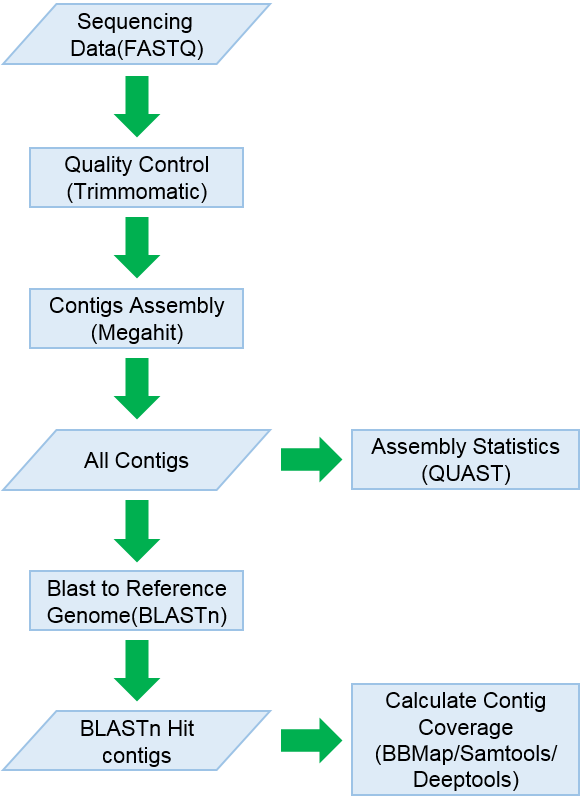
'De novo-Assembly' can be used to assemble raw NGS sequencing data, compare the assembly contigs to SARS-CoV-2 reference database, identify SARS-CoV-2 genetic sequences from the assembly contigs, quality evaluation of assembly contigs, analyze the degree of contig coverage, sequencing depth, etc.
Reference running time is tested using real data set when the server system is idle (not including upload time). Running time of actual tasks depend on the workload state of the server system, data volume, data quality and so on.
| Data1 | Data2 | Data3 | Data4 | Data5 | Data6 | |
| SINGLE/PAIRED | SINGLE | PAIRED | PAIRED | PAIRED | PAIRED | PAIRED |
| Data volume | 118Mb | 203Mb | 1.0Gb | 1.5Gb | 2.2Gb | 8.0Gb |
| Calculating time* | 1m34s | 1m48s | 23m12s | 41m18s | 1h5m28s | 41m12s |
| Accession | SRR11247077 | SRR10903402 | SRR11092064 | SRR11092057 | SRR11092058 | SRR10971381 |
*Run on 10 Threads, 50G Memory