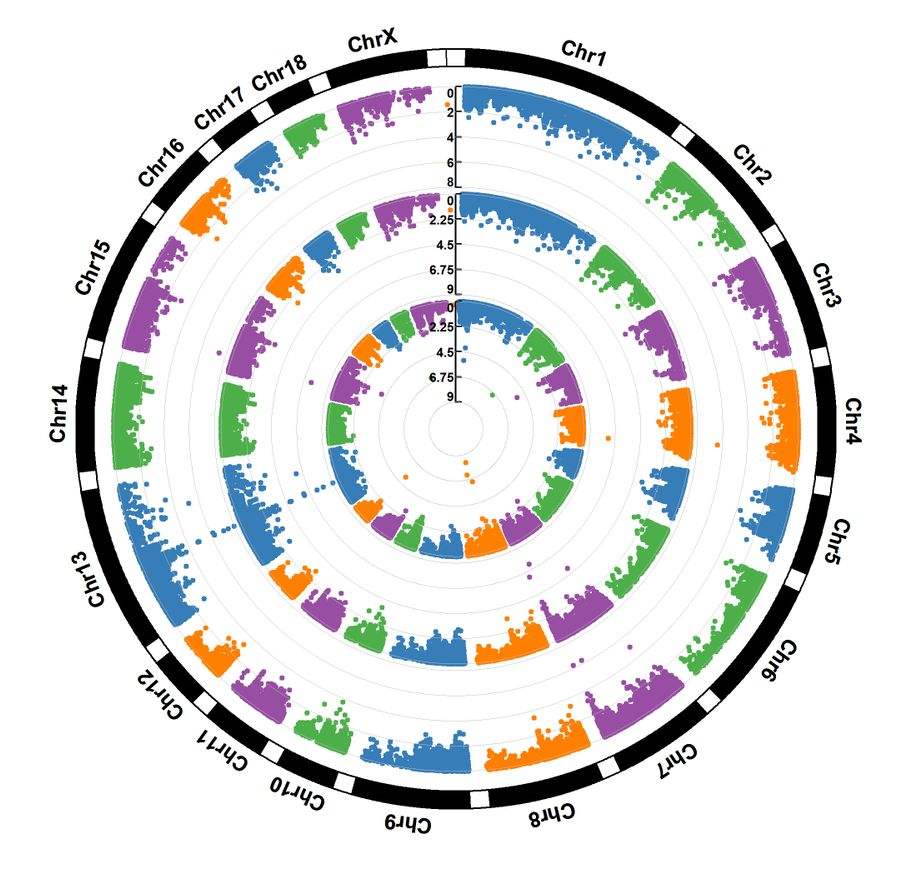
 easyGWAS
easyGWAS
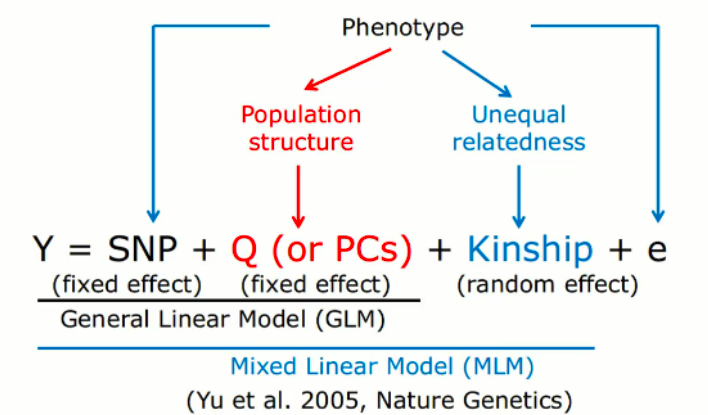
When uploading your own genotype dataset, please use the block-gzipped vcf file, with MAF and missing rate filtered. As it can compress your input file to a reasonable size.
A one-line pipeline is recommended:
vcftools [–vcf <your_vcf_file>|--gzvcf <your_vcf.gz_file>] --maf 0.05 –max-missing 0.9 –recode –out | bgzip –c > output.vcf.gz
(This pipeline deals with vcf/vcf.gz file to discard the sites with maf < 0.05 and missing rate > 0.1, then change it to vcf.gz file)